We are excited to share our publication (Jennings et al. 2025) in Cardiology in the Young describing discussion needs and potential research avenues for young people with dilated cardiomyopathy (DCM). We hope it is a valuable insight for clinicians and researchers, particularly for the ongoing development of clinical care specifically for the young with DCM.
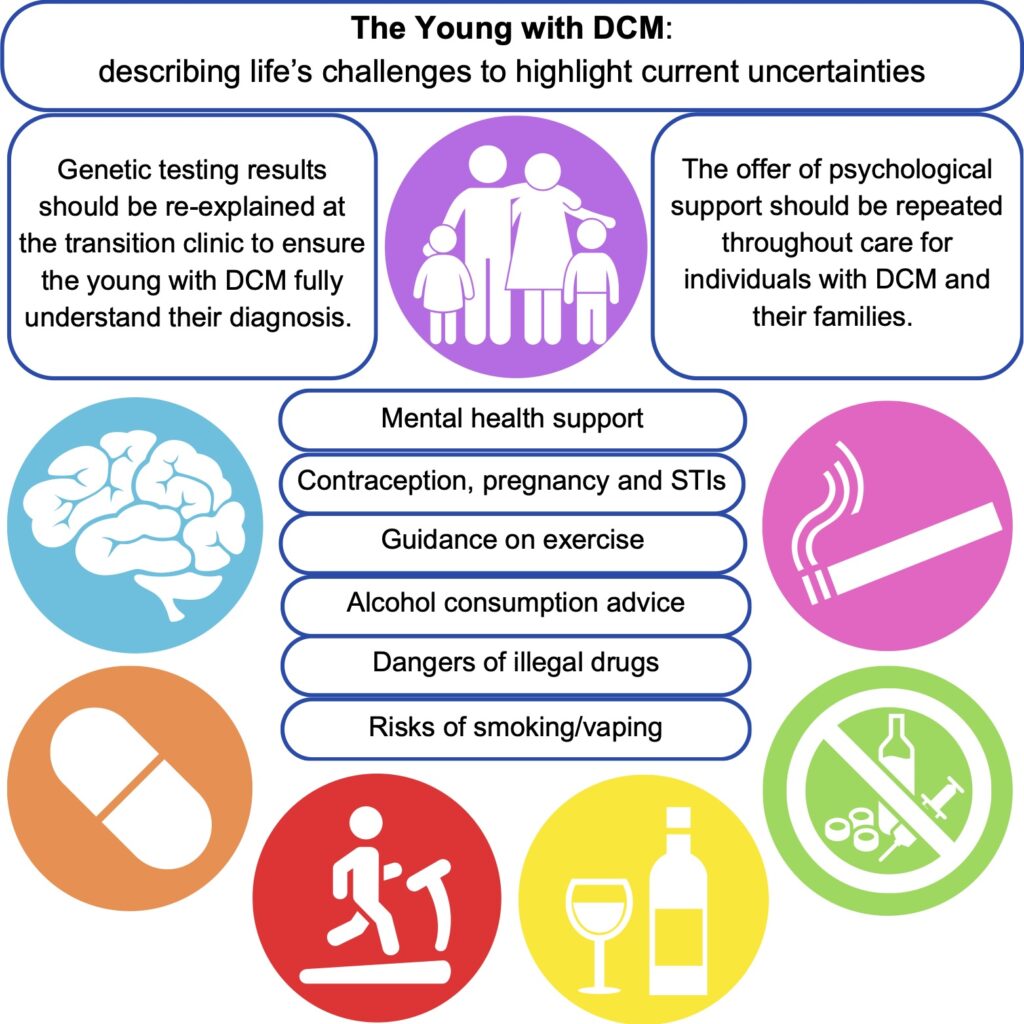
DCM is a heart condition characterised by an enlarged left ventricle that can lead to heart failure. It is most often diagnosed in adult males, however, when diagnosed in childhood, it can present unique challenges to patients and clinicians. As children move from the paediatric hospital services to the adult services (“transition”), they need unique support when taking responsibility for their health and living with DCM.
Using a patient-scientist’s perspective to inform a literature review, we identified areas that could form the basis of the information provided during transition clinical services. This could aid the further standardisation of clinical care for young people and ensure that the correct support is in place to equip young people to make informed decisions and provide an opportunity for discussion about topics that may be experienced when living with a chronic condition. The key areas identified as requiring additional support from clinical teams are: mental health, exercise, alcohol consumption, cigarettes, recreational drugs, and sexual health.
Young people with DCM and their families require specialised mental health support to equip them for lifelong clinical care. For patients, it is important to support them as they come to terms with their limitations amongst healthy peers. Family members may have experienced traumatic events such as sudden death or feel the empathetic burden of being the “well” sibling. Medical trauma during childhood can impact stress tolerance in later life and young people may be more susceptible to poor mental health outcomes without intervention. Repeated signposting to mental health services is recommended for both the patient and their families.
Genetic testing is often undertaken for diagnostic support in cardiomyopathies. If carried out during childhood, results should be revisited with the young person in the transition to adulthood. This would ensure the patient has a full understanding of the downstream effects of a given result and the impact on access to family planning measures, etc.
Current exercise guidelines suggest it is safe to exercise to perspiration with the ability to hold a conversation and avoid exercising when feeling unwell or tired. The advice however is generally non-specific regarding varying exercise types and duration, with little research available on DCM in the young.
Alcohol and caffeine are regularly consumed in adults, but the evidence surrounding their negative impacts is non-specific, and the advice is to avoid them in excess, hydrate more before and after consumption, and avoid taking them in combination. They may oppose the effects of therapeutic drugs used for DCM treatment.
Smoking, vaping, cannabis, cocaine, MDMA, and ketamine, negatively influence the cardiovascular system, with cocaine described as the most cardiotoxic. There is little specific evidence for patients with cardiomyopathy, and it is recommended by medical professionals to avoid consumption. When advocating abstinence to teenagers, it is important to discuss the reasoning for this.
Pregnancy can be contraindicated in some people with DCM, and progesterone-only or barrier methods of contraception are recommended to avoid the risks associated with oestrogen-based alternatives. During the transition to adult services, the implications and planning of pregnancy should be discussed to ensure that patients are well-informed about the importance of avoiding unplanned pregnancies.
Further research is needed to address the many uncertainties in these areas with respect to young age, particularly for physical activity, and such guidance would be welcomed by the young with DCM who must come to terms with being different and more limited amongst healthy peers.