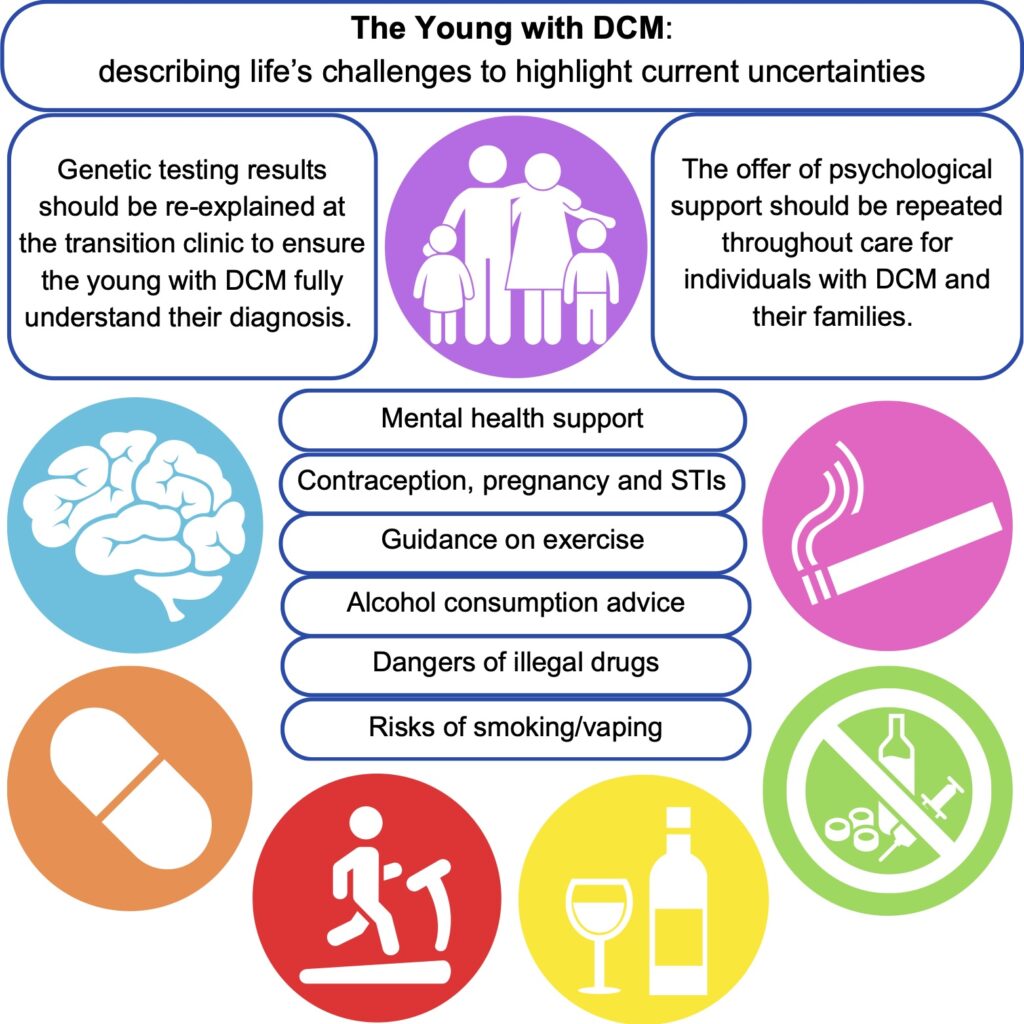
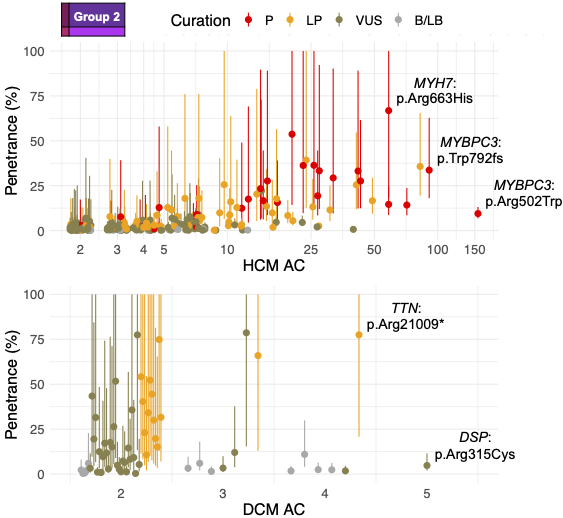
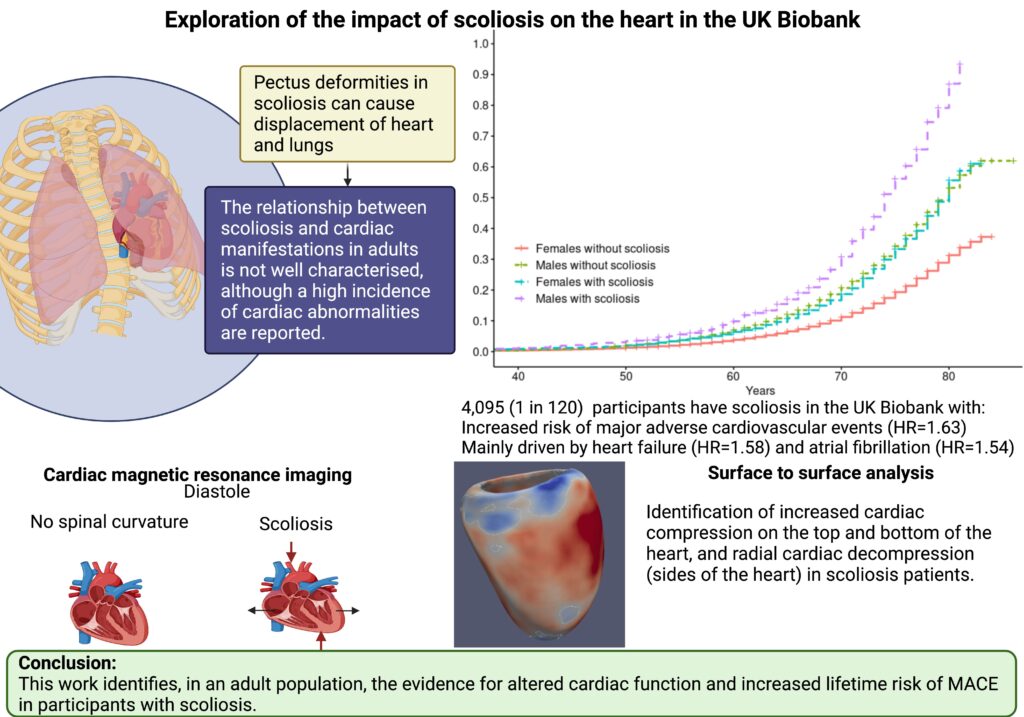
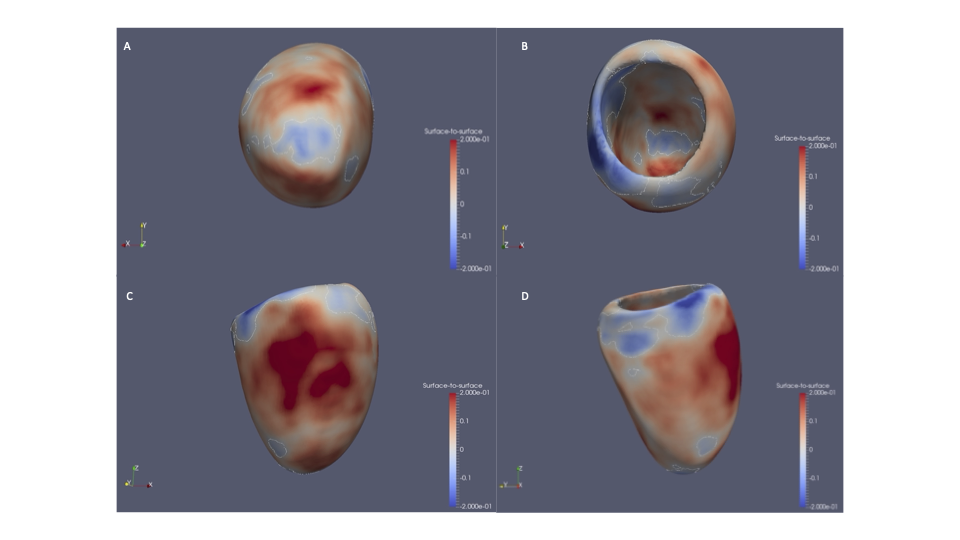
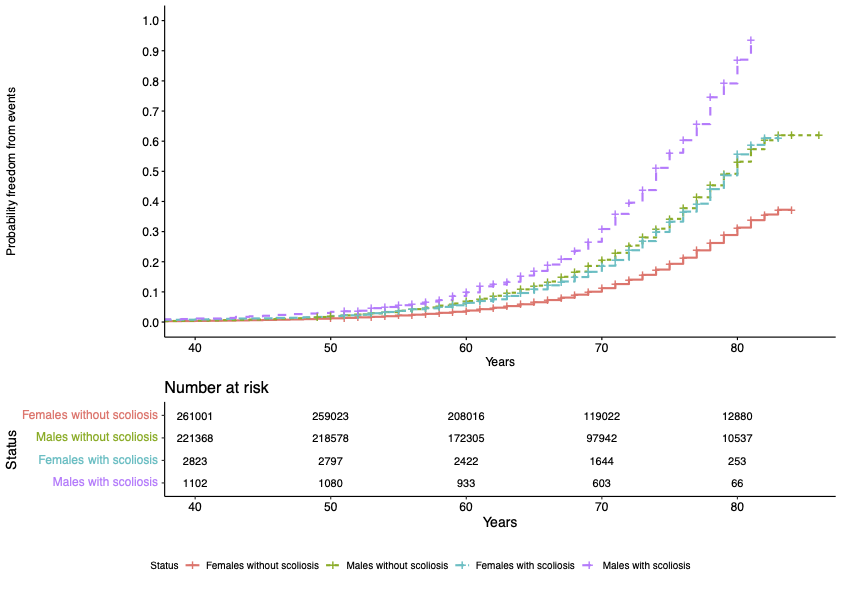
Dr Kathryn McGurk is a geneticist and data scientist at Imperial College London. Her research focuses on understanding how genetics affects cardiomyopathies and heart structure and function, and she has a special interest in takotsubo.

What is Takotsubo?
Takotsubo is a sudden and severe heart condition. While under-recognised, it affects 1 in 20 women suspected of a heart attack and men too. It mainly affects women over 65 years old. It is no longer thought to be a cardiomyopathy, but an “acute myocardial ischaemic syndrome”.
The symptoms of a takotsubo episode closely resemble a heart attack: chest pain, breathlessness, ECG reading changes and biomarkers. Patients are rushed into treatment for an emergency heart attack. However, unlike a heart attack, takotsubo is not caused by a blockage in the main blood vessels of the heart. A takotsubo diagnosis is eventually made through the exclusion of other conditions; this is not good enough for patients or clinicians.
Takotsubo is very poorly understood. It is thought to happen to some people when their body is coping with overwhelming physical or emotional stress, but this is not always the case and has not been studied well. The hallmark of takotsubo compared to heart attacks is the “recovery” of heart function within weeks. However, this “recovery” is only partial; recurrence of takotsubo is common, and many people have long-term problems after the first hospital visit.
Why are you interested in studying takotsubo?
A few years ago, I attended Cardiomyopathy UK’s annual patient conference in London. There, I met a group of individuals diagnosed with takotsubo, who noted that they didn’t have an emotional trigger or stress event. They wanted to be involved in more research to better understand their condition.
Once I started studying takotsubo, I was surprised at the lack of understanding and research for this common condition. We do not know why it happens, why it mainly affects women, or which patients are most at risk of complications. There are no specific treatments or preventative measures. No therapies have been shown to reduce recurrences or any other major cardiovascular events. There is no national picture of how this is managed across the NHS.
I think we can better understand what causes takotsubo in our research group with the help of people who are affected.
What research projects are you currently working on?
My current takotsubo research is split into two parts – understanding current NHS care pathways and outcomes, and using genetics as a tool to guide biological understanding. I am also working with a fantastic group of experts, the MINOCA workstream, as part of the NIHR-BHF Cardiovascular Partnership. This UK-wide collaboration will improve our understanding of takotsubo in the near future.
Part one: understanding current NHS care pathways and outcomes
Right now, we are analysing data from the NICOR Myocardial Ischaemia National Audit Project (MINAP), a nationwide NHS database that records every hospital admission for suspected heart attacks in England and Wales. Since many takotsubo patients first appear as suspected heart attack cases, MINAP contains one of the largest collections of real‑world information on this condition (over 5,000 individuals). Uniquely, clinicians were asked to fill in a voluntary discharge questionnaire when the patient was diagnosed with takotsubo. We have access to the data from this questionnaire, and it includes information since 2013 on over 2,000 individuals diagnosed with takotsubo.
Using this unique national dataset, we will examine:
- What kinds of emotional or physical stress triggers are most common, and are they likely a reason for this diagnosis, or are they as common in the population?
- Which patients are most affected (e.g., age, sex, medical history)?
- Symptoms and heart scan findings.
- How patients are treated in hospital, including medications and specialised cardiac support?
- How often do serious complications occur, such as abnormal heart rhythms or blood clots?
- How many patients experience repeat episodes and have a family history of takotsubo?
- How do takotsubo patients differ from other patients admitted with suspected heart attacks?
The results will help clinicians recognise takotsubo more quickly, avoid unnecessary invasive procedures, and provide more personalised care. This will lay the groundwork for our future research into the biological causes of takotsubo, including genetics and imaging studies, to provide targets for prevention and treatment. Ultimately, we aim to improve the outcomes for thousands of people each year who experience this misunderstood condition and are initially managed for the wrong condition.
Part two: using genetics as a tool to guide biological understanding
Genetics may help us identify the cause of takotsubo and target therapeutics for this condition if it is similar to other cardiovascular conditions. We know that most of the genes involved in keeping the heart beating are in the left ventricle, and these genes can influence someone’s risk of cardiomyopathy. Do the same genes influence the risk of takotsubo? This understanding will help us find genetic markers to identify people who are at risk of takotsubo much earlier.
The NHS database we are using in “part one” does not include genetic information, so to do this, we are inviting people who have been diagnosed with takotsubo to join “The Heart Hive Cardiomyopathy Study”.
I have takotsubo. How can I get involved?
We are inviting takotsubo-diagnosed individuals and their family members to join the Heart Hive – an online portal where people can sign up to participate in research studies (thehearthive.org).
We will post takotsubo studies, and those who have signed up can get involved if they like. This may include opinions on future research plans, surveys, or participation in genetic studies.
“The Heart Hive Cardiomyopathy Study” is already running on the Heart Hive and will help us better understand takotsubo compared to other cardiomyopathies. Taking part in this study involves providing a saliva sample by post for DNA sequencing. This will help us finally understand takotsubo through better research.
Steps to register and fill out the survey:
Website: https://thehearthive.org/
Register: https://thehearthive.org/studies/hh_registry/join/preEnroll
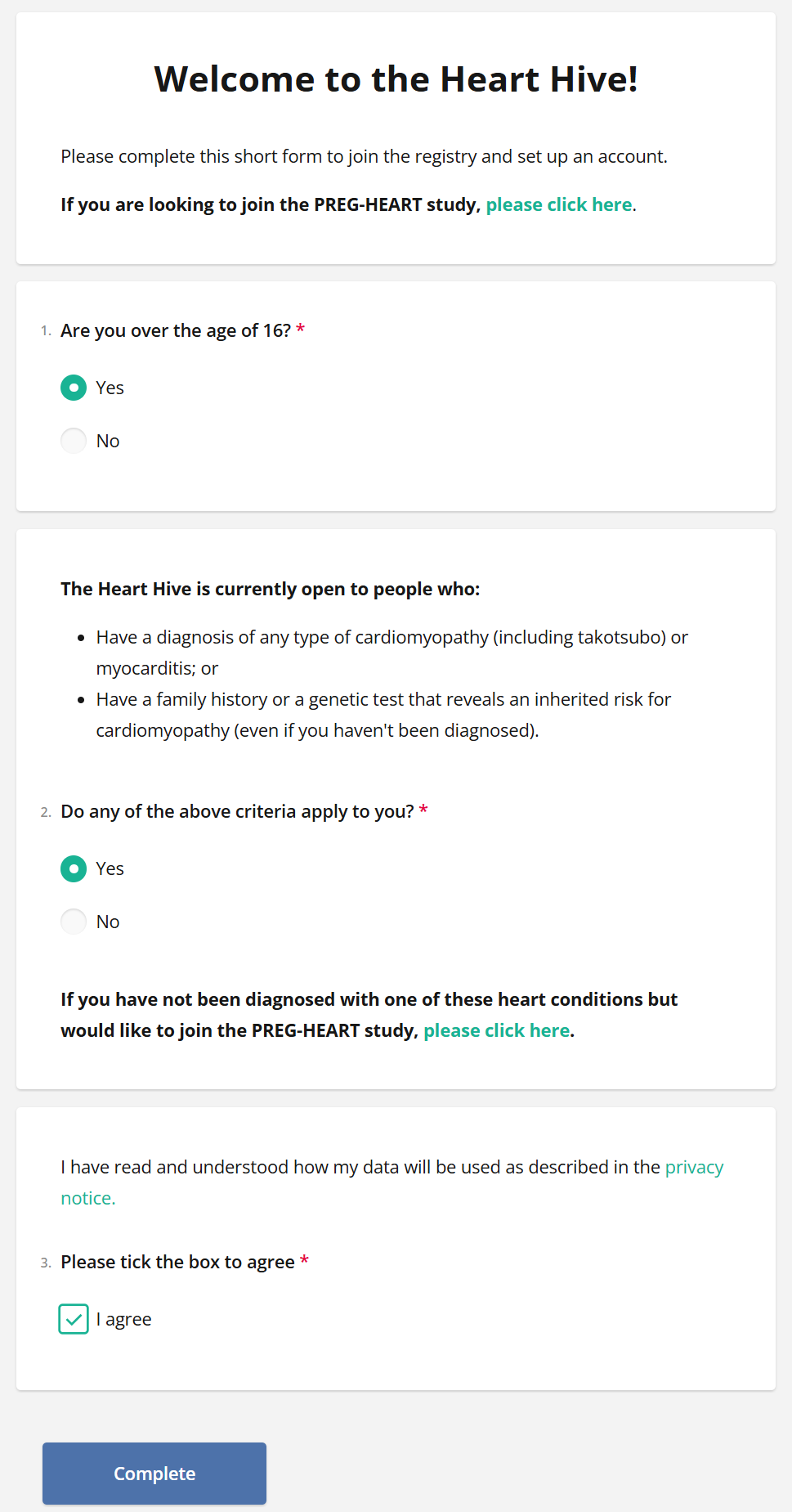
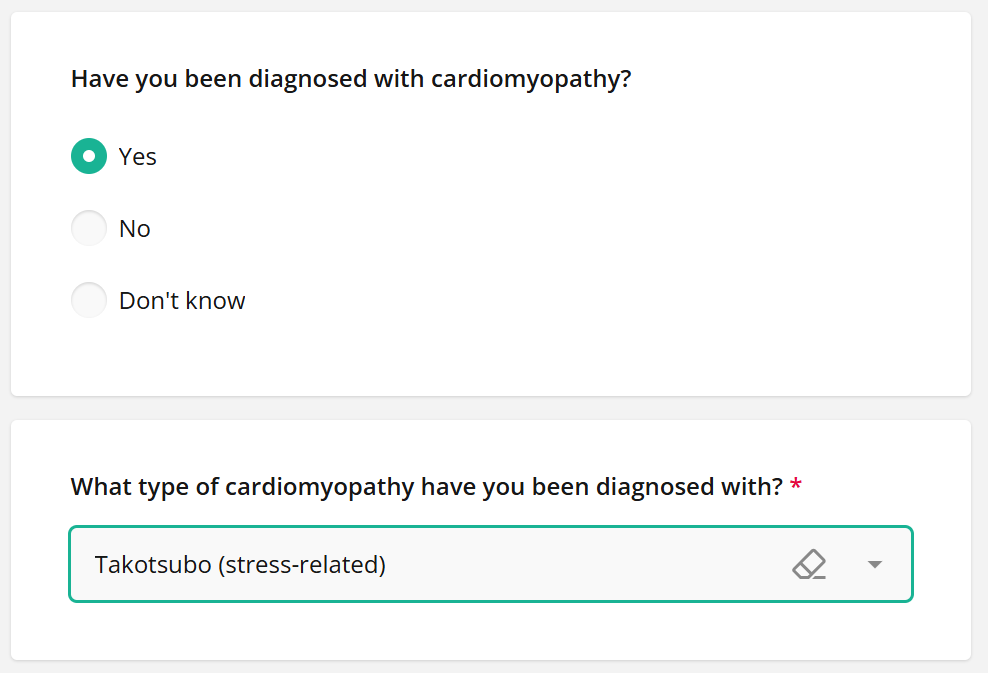
We have categorised takotsubo under cardiomyopathies,
so say “Yes” if you or a family member has been diagnosed
and you’d like to be notified of research opportunities:
Next is email verification and consent to contact:
Then, register! We’ve categorised takotsubo under cardiomyopathies:
The most time-consuming part is the Basic Health Information survey; your first survey that can be part of research! This information will be provided to the studies that you would like to be involved in. This information is really useful to researchers and clinicians trying to understand takotsubo.
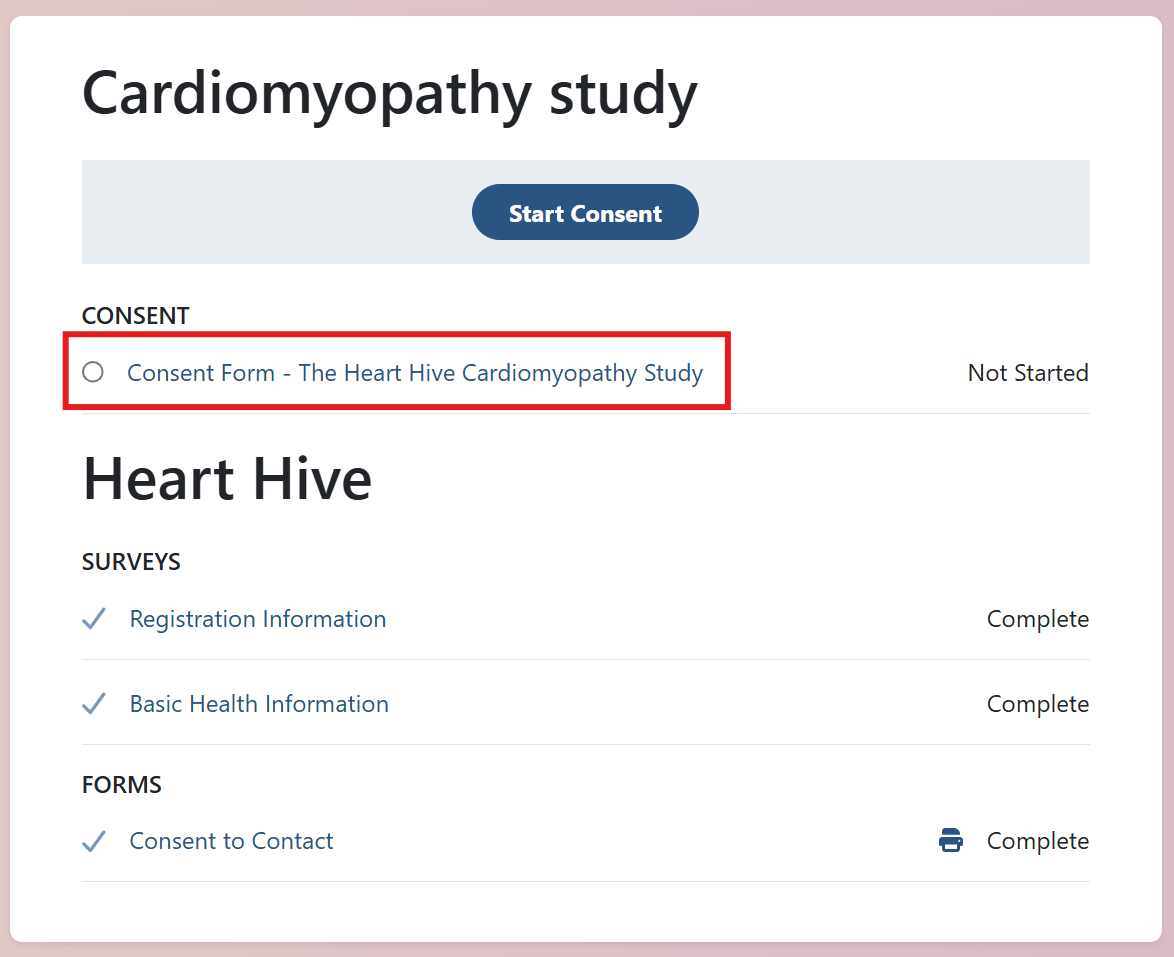
All set! You’re now in the Heart Hive portal and will be notified of future research opportunities.
There is a study already posted looking at genetics of takotsubo in the UK, please do get involved:

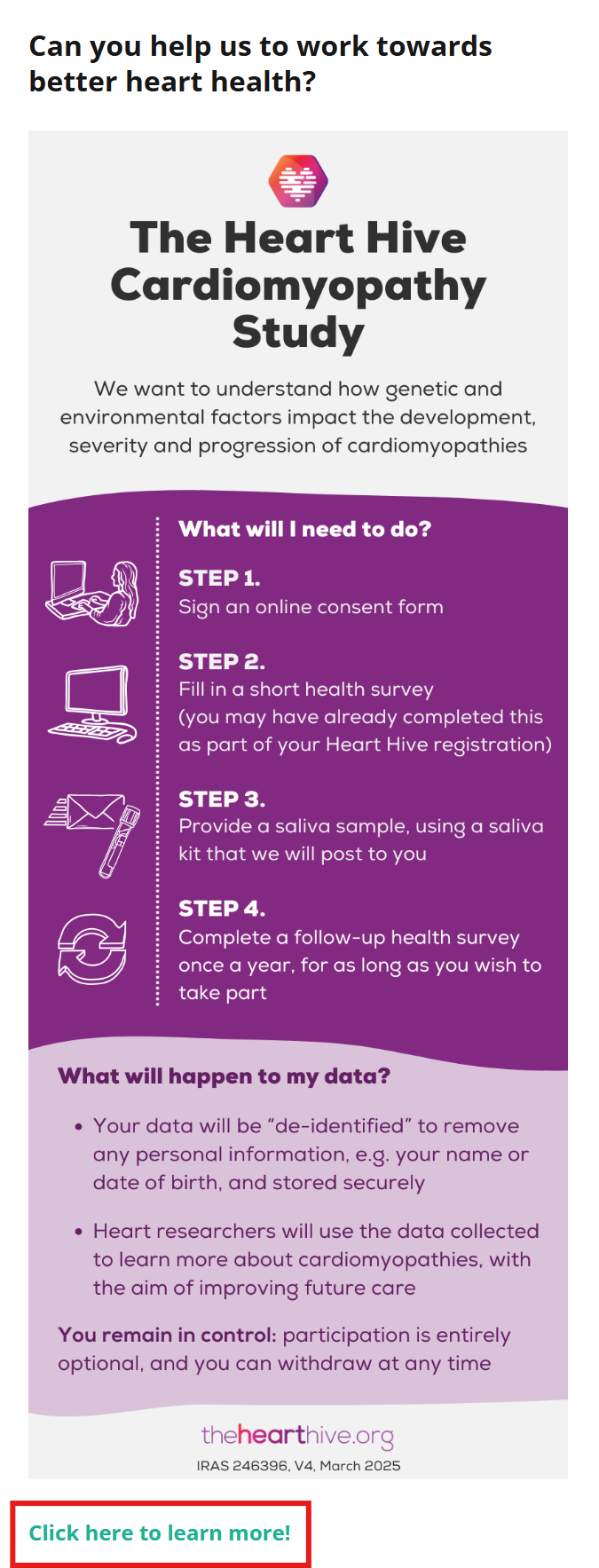
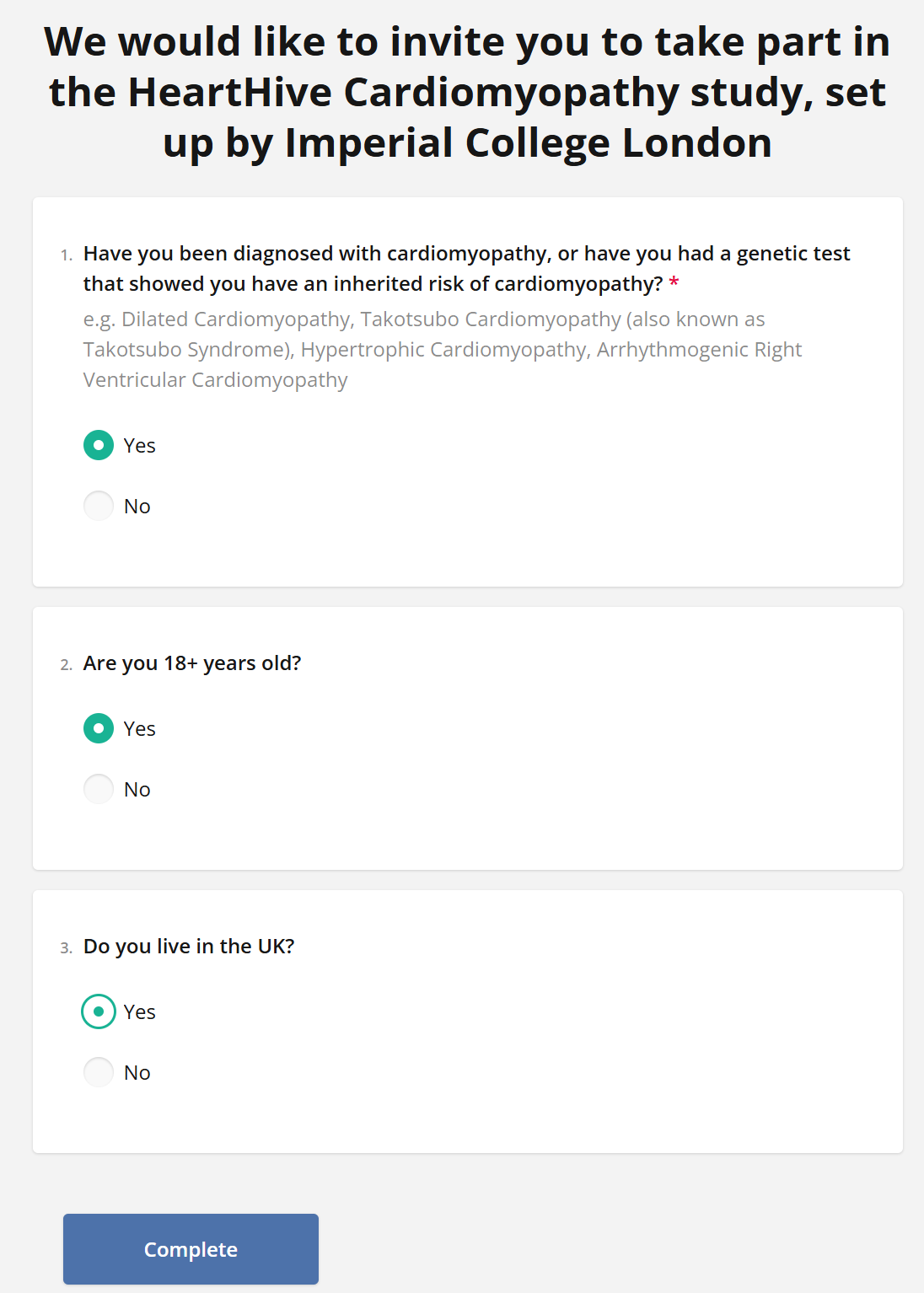
The study is led by Professor James Ware, who is the head of our cardiovascular genetics research group and is supporting my goals of gaining a better understanding of takotsubo: