Rare and extremely rare variants can be difficult to interpret, and at times genotype-phenotype associations can be hard to determine. DECIPHER is an interactive web-based database of genomic variants and associated phenotypes. It contains data from an international community of more than 270 centers and academic departments that focus on clinical genetics and rare disease genomics.
The web interface allows for depositing and sharing of genomic variants and phenotypes associated with an individual patient. Centers that deposit a variant can choose to keep data private, share with other members within a pre-defined consortium, or with patient consent can contribute to the open-access data that is available on DECIPHER.
DECIPHER was established in 2004 to enable the interpretation and sharing of genotype and phenotype data from rare disease patients. The interpretation of cardiac/cardiovascular disease is complex as these are late-onset, variable penetrance conditions. We have been collaborating with DECIPHER to implement features to enhance the interpretation of cardiac/cardiovascular disease, including the incorporation of additional data and providing a more intuitive experience for cardiac users, specifically when exploring the impact of a particular cardiac variant. In this blog post we describe how cardiac users can utilize the DECIPHER platform, with particular focus on these recent updates.
Flexible search bar
Within the cardiac community, different users (clinicians, diagnostic labs or researchers) may have their variant of interest formatted using a variety of positional identifiers. DECIPHER can now accept variants as search terms in a variety of formats.
- Users can identify a gene by HGNC symbol, MANE select transcript or Ensembl identifier
- Search bar can take the position within the gene as a coding DNA reference, protein reference, or using a “gnomad-style” search based on chromosome and genomic coordinates
- Reference and alternate alleles can be identified using 1 or 3 letter amino acid codes
- Additionally, the input bar allows for space or colon separators, and optional “p.” and “c.” prefix for cDNA and protein reference location
The result of incorporating these options allows for a wide range of search terms that a user can input to access data about a variant of interest. Most formatting used across the wider cardiac community is now accepted.

Variant summary table
The landing page after entering a search term now leads with a summary that indicates whether the variant has been seen across multiple cohorts and datasets. Quick access to information about the presence of the variant in healthy or disease patients via DECIPHER patients, Deciphering Developmental Disorders (DDD) research variants, ClinVar, gnomAD, and other disease cohorts now including Cardiac VariantFX, can be informative as evidence towards a variant’s pathogenicity.

Cardiac VariantFX Case/Control frequencies
Cardiac VariantFX has collected sequence and variant data from hypertrophic cardiomyopathy patients, dilated cardiomyopathy patients and healthy controls from a total of 7 centres*. Presence or absence of the variant in cardiac disease cohorts and/or confirmed healthy controls can be informative when assessing pathogenicity of a variant that is potentially associated with cardiac disease.
Only variants in genes known to be associated with hypertrophic or dilated cardiomyopathy at the time of processing are available on DECIPHER (ACTC1, BAG3, DES, DSP, FLNC, LMNA, MYBPC3, MYH7, MYL2, MYL3, PLN, RBM20, SCN5A, TNNC1, TNNI3, TNNT2, TPM1, TTN). Note that not all genes are sequenced in each cohort.
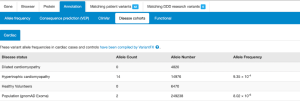
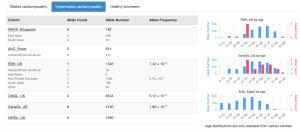
Gene-disease associations supported by cardiac cohort data
The landing page for each gene or variant search is a Gene-disease associations page (found below the variant summary table). For each gene, there are a number of sources (OMIM, PubMed Gene Reviews, Gene2Phenotype, ClinGen and GenCC) which have investigated conditions that are potentially associated. Associations listed on this page are not always definitive, and some investigations have concluded a lack of association.
In addition to the sources mentioned above, DECIPHER now also displays associations under a “Case/Control cohort data” heading, which now includes disease associations which have been supported by data included in the cardiac VariantFX cohort data.
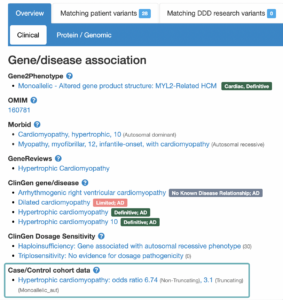
Gene-disease association statistics
Clicking on the association supported by Case/Control cohort data opens a window that contains statistics that have been calculated based on cardiac cohorts** which support the association. Statistics are calculated individually for Truncating variants, Non-truncating variants, and All variants combined.
This window also contains a shortened text version of the mechanism narrative found in G2P. The full text version of the mechanism narrative can be found via the G2P VEP plugin.

PM1 regions browser
A variant located in a mutational hotspot or well-established functional domain is considered moderate evidence of pathogenicity (PM1, Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology ).
Disease associated genes have been assessed for PM1 (hypertrophic cardiomyopathy and dilated cardiomyopathy), and the identified regions are now shown in a track of the DECIPHER protein browser.

* Royal Brompton and Harefield Hospitals in London, UK, Aswan Heart Center in Egypt, National Heart Centre Singapore in Singapore, Laboratory for Molecular Medicine of Partners HealthCare Personalized Medicine in Boston, US, Oxford Regional Genetics Laboratory in the UK, GeneDx in Maryland, US, and Northern Ireland Regional Genetics Laboratory in Belfast, UK
** Excess in Disease, Aetiological Fraction, and Odds Ratio
References
Walsh , et al. Genet Med. 2017 Feb;19(2):192-203. doi: 10.1038/gim.2016.90. Epub 2016 Aug 17. PMID: 27532257; PMCID: PMC5116235.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, Ahmad M, Mazaika E, Allouba M, Zhang X, de Marvao A, Day SM, Ashley E, Colan SD, Michels M, Pereira AC, Jacoby D, Ho CY, Thomson KL, Watkins H, Barton PJR, Olivotto I, Cook SA, Ware JS. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019 Jan 29;11(1):5. doi: 10.1186/s13073-019-0616-z. PMID: 30696458; PMCID: PMC6350371.
Morales A, Kinnamon DD, Jordan E, Platt J, Vatta M, Dorschner MO, Starkey CA, Mead JO, Ai T, Burke W, Gastier-Foster J, Jarvik GP, Rehm HL, Nickerson DA, Hershberger RE; DCM Precision Medicine study of the DCM Consortium; Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med. 2020 Apr;13(2):e002480. doi: 10.1161/CIRCGEN.119.002480. Epub 2020 Mar 11. PMID: 32160020; PMCID: PMC8070981.
Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, Jongbloed JDH, Macaya D, Manrai A, Orland K, Richard G, Spoonamore K, Thomas M, Thomson K, Vincent LM, Walsh R, Watkins H, Whiffin N, Ingles J, van Tintelen JP, Semsarian C, Ware JS, Hershberger R, Funke B. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018 Mar;20(3):351-359. doi: 10.1038/gim.2017.218. Epub 2018 Jan 4. PMID: 29300372; PMCID: PMC5876064.