Dr. Nicky Whiffin
Today we are very pleased to announce the release of CardioClassifier (cardioclassifier.org), a web-tool to support interpretation of variants identified in genes associated with inherited heart conditions.
In this blog post we describe a bit about the project, our motivation for creating the tool, and how you can get started.
Why did we create CardioClassifier?
Genetic testing for inherited heart conditions is commonly available in the clinic. It can be used to help diagnose disease, direct patient management and for screening in relatives. Although DNA sequencing is now relatively routine and straightforward, the major challenge in genetic testing remains interpreting the identified variants.
Why is variant interpretation such a challenge?
The genetics of inherited heart conditions is complex with multiple genes implicated in each disorder. Although there are a few recurrent variants that are identified in multiple cases, disease-causing variants are often unique to one individual patient (and their family). As each one of us carries thousands of genetic variants, identifying which of these have the potential to be disease causing can be a challenge.
So how do you even start?
Interpreting a genetic variant can feel a bit like a forensic investigation; we ask a lot of questions and collate all the evidence we can find. Has this variant been seen before? If yes, was this in a patient with the same disease or in healthy individual(s)? Can we predict what effect this variant has on the protein? Does the variant reside within a region of the protein known to perform an important function, or where we commonly see disease mutations? Is this variant seen in all individuals with the disease in the family?
In January 2015, the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) jointly released a set of guidelines aiming to standardise this process [1]. They outlined exactly which questions we should be asking (28 to be exact) and how much weight we should give to each answer.
Although these guidelines were a huge step forward, they were developed to apply to any variant in any gene and in any disease. In reality, the exact questions we ask and the exact places we need to look for the answers are highly disease dependent, and as such, the guidelines need to be curated for each disease.
But doesn’t this all take a lot of time?
You might be thinking that this all sounds like an awful lot of work, and you would be right; interpreting a variant requires collecting data from a multitude of different sources and integrating this all into a final decision. In fact, this is what clinical scientists in genetics labs across the world do every single day.
So where does Cardio Classifier fit in?
We set out to make a bioinformatic tool that would make the process of variant interpretation both easier and better. By adding in some automation we can standardise the process and collect a lot of the data required to interpret a variant all in one place.
Specifically, the tool aims to do the following:
- Reduce the time taken to interpret each variant by aggregating data from multiple different sources
- Incorporate expertly curated disease and gene-specific knowledge, data and thresholds
- Use the aggregated data and disease specific thresholds to automatically assess each variant against ACMG/AMP criteria
- Be interactive to allow an interpreter to add case-level data and thereby refine a classification
- Be fully transparent so that an interpreter can see exactly what evidence has gone in to reaching a final classification
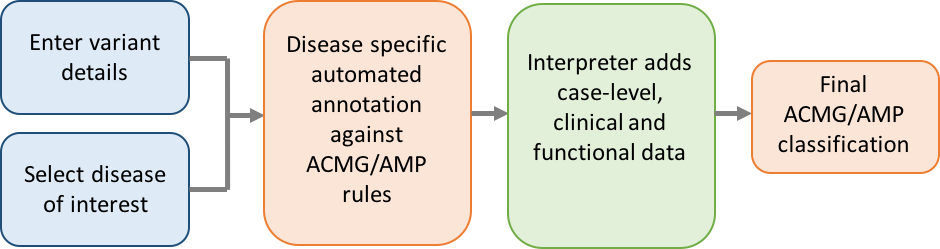
A schematic showing the workflow of variant interpretation using CardioClassifier
Great, so how do I get started?
Step 1 – register for an account
Head on over to cardioclassifier.org, click ‘REGISTER’ and enter your details. After accepting the licensing terms you will be sent an email to confirm your email address (please check your spam) before your account will be activated.
CardioClassifier is freely available for the NHS and for research,. For other healthcare or commercial use (including UK private practice) please contact cardioclassifier@gmail.com
Step 2 – enter variant details
Once you click ‘LOG IN’ you should be directed to this variant upload page:

A screen shot of the variant upload page
In the left side of the box, (i) add a sample ID of your choice (NB do not use patient identifiable information) (ii) choose from a list of 11 diseases (the disease you select dictates both the genes analysed and the disease/gene specific thresholds used).
In the middle section add your variant details. You can either upload a single-sample variant call file (VCF) by selecting ‘Choose file’ and browsing on your computer, or you can enter variant details in the box below.
If you are stuck for something to try for now, enter this example into the box ‘chr14 23894969 C A het’ and select ‘Hypertrophic Cardiomyopathy’ as the diagnostic test.
Click on ‘Create Report’ and wait for the magic to happen!
What is paralogue annotation?
The upload page gives you the option to include ‘paralogue annotation’. If you select this option, your variant(s) will be assessed against two new criteria utilising known disease-causing variants in related genes/proteins (paralogues) [2]. Full details can be found in our documentation (link on the variant upload page). Since this evidence is not currently included in the ACMG framework you need to opt in to include it.
Why can I not search for any gene and/or disease?
We have invested significant effort into defining if each individual rule should be assessed, and what threshold should be used, for each individual gene and disease pair. We have therefore focussed on 40 genes with robust links to 11 diseases and genes where we are confident we know something about the mechanism through which they cause disease. We will expand the list of genes as more data becomes available. Details of disease and genes included are in the tool documentation (link on the variant upload page).
Step 3 – automated variant annotation
For those of you interested in the detail, the tool proceeds as follows:
- The uploaded VCF/variant details are filtered to only those in our 40 genes
- Variants are run through the Ensembl Variant Effect Predictor
- An extensive database is used to annotate each variant against up to 17 ACMG/AMP rules which can be computationally predicted
- Each variant is compared to extensive datasets of population controls, healthy controls and disease cohorts (including over 7,000 cardiomyopathy cases)
- An interactive report is printed out using PHP
Step 4 – interact with the report
For each variant, a box is printed out that looks a little like this:

A screen shot of the variant output
The top half of the box is a representation of the grid from the ACMG/AMP guidelines with each rule that has been activated for this variant shown in colour. Boxes with a grey background represent rules that cannot be computationally predicted and that require user input. This grid is fully interactive – click on a box to add or remove it as evidence.
The bottom half of the box displays the evidence that went in to assessing each of the ACMG/AMP rules, including case and control frequencies and links out to various external resources (PubMed, Google, ClinVar, UCSC, ACGV, ExAC and the Beacon Network).
To complete the classification of your variant(s), follow the links out to publications and ClinVar to find any reported clinical or functional information. Use this, along with any information you have about your specific patient/sample, to activate any rules that have not been activated automatically, by simply clicking on the rule you want to activate. When activating (or deactivating) a rule you are given the option to include a comment – anything you add in this box will appear at the bottom of the variant report under ‘Manually added data’ as additional evidence. Each time a rule is adjusted, the overall variant classification in the top left of the box is re-calculated according to the ACMG/AMP logic.
The data you add at this step is not currently stored, however, the ability to save and return to previous annotations is the first in our long list of features to add to the tool!
Getting help
We have included some more instructions, including file format requirements and details of how we have parameterised the rules in our documentation. There is a link to this on the file upload page. If you are still stuck, or you find a bug, please email us on cardioclassifier@gmail.com.
Building a knowledge-base
Our aim is to make variant classification with CardioClassifier as easy as possible and constantly adapt it to include new data as it becomes available. As expert groups, such as the ClinGen resource (www.clinicalgenome.org), release guidelines specific to individual genes or diseases, we will build these into the tool. We will also be adding case-level and functional data for a set of highly curated variants. As mentioned above, in the near future we will add functionality to save the annotations you add interactively, so that when you return to the same variant again, you do not have to repeat all of your hard work. In these ways we hope that CardioClassifier will become a highly curated knowledge-base for variant interpretation in inherited heart conditions.
Acknowledgements
Developing and releasing CardioClassifier has been a huge team effort by the Cardiovascular Genetics and Genomics team at Imperial College London, the Royal Brompton Hospital and the Medical Research Council London Institute of Medical Science. We are also grateful to our funders; the British Heart Foundation, the Wellcome Trust and the Department of Health.
References
- Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24
- Ware JS et al. Paralogous annotation of disease-causing variants in long QT syndrome genes. Hum Mutat. 2012 Aug;33(8):1188-91
Save
Save
Save
Save
