We are pleased to share our latest publication in Circulation on the genetic architecture of acute myocarditis and the overlap with inherited cardiomyopathy.
Myocarditis refers to inflammation of the heart muscle. It is traditionally considered to be a random event, which may be triggered by various infections, autoimmune conditions or toxic insults. Whilst most patients show spontaneous recovery, a small but important subset can suffer with long-term adverse cardiac events, including the need for heart transplantation or cardiac device implantation (1).
The purpose of this study was to answer a key question – is there an underlying genetic susceptibility that may explain the wide heterogeneity in clinical outcomes seen with acute myocarditis.
To address this, we compared 3 main cohorts:
– Cohort 1: 230 patients recruited consecutively in London (UK) presenting with acute myocarditis confirmed on cardiovascular magnetic resonance (CMR) or myocardial biopsy (2).
– Cohort 2: 1053 community based healthy volunteers in London with no history of cardiovascular disease and a normal CMR scan.
– Cohort 3: 106 patients presenting with acute myocarditis confirmed on myocardial biopsy in Maastricht (Netherlands).
All participants underwent targeted DNA sequencing for well-characterized cardiomyopathy-associated genes with comparison to healthy controls sequenced on the same platform. The primary outcome was all-cause mortality.
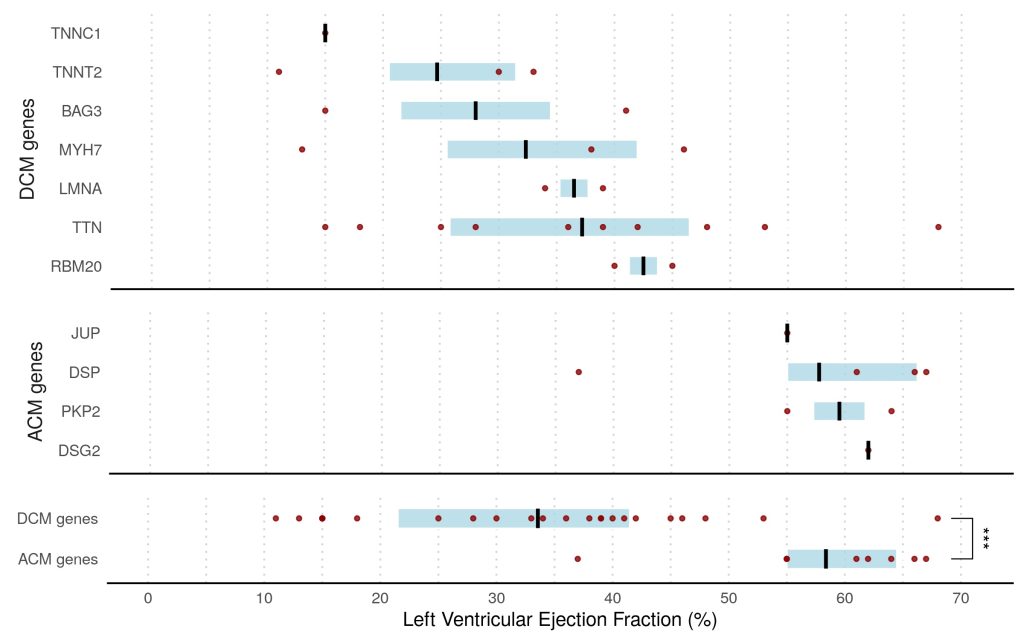
Overall, we found that 8% of patients presenting with acute myocarditis (27 out of 336 cases) carried likely pathogenic variants in ACM or DCM associated genes compared to <1% in healthy volunteers (p<0.0001).
This finding was dominated by truncating variants in Titin (TTN) in 7% of patients, all with left ventricular ejection fraction <50%, compared with 1% in controls (odds ratio, 3.6; P=0.0116). ACM-associated genes were found in 3% of cases versus 0.4% of controls (odds ratio, 8.2; P=0.001). This was driven predominantly by truncating variants in desmoplakin (DSP) in patients presenting with chest pain and preserved LV ejection fraction.
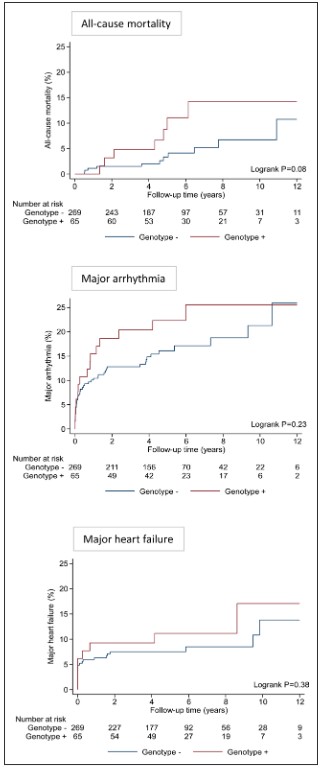
Over a median follow-up of 5.0 years (IQR, 3.9–7.8 years), there was a trend toward greater all-cause mortality in genotype-positive patients compared with genotype-negative patients (5-year mortality risk 11.1% vs 3.3%; P=0.08 after adjusting for age and sex).
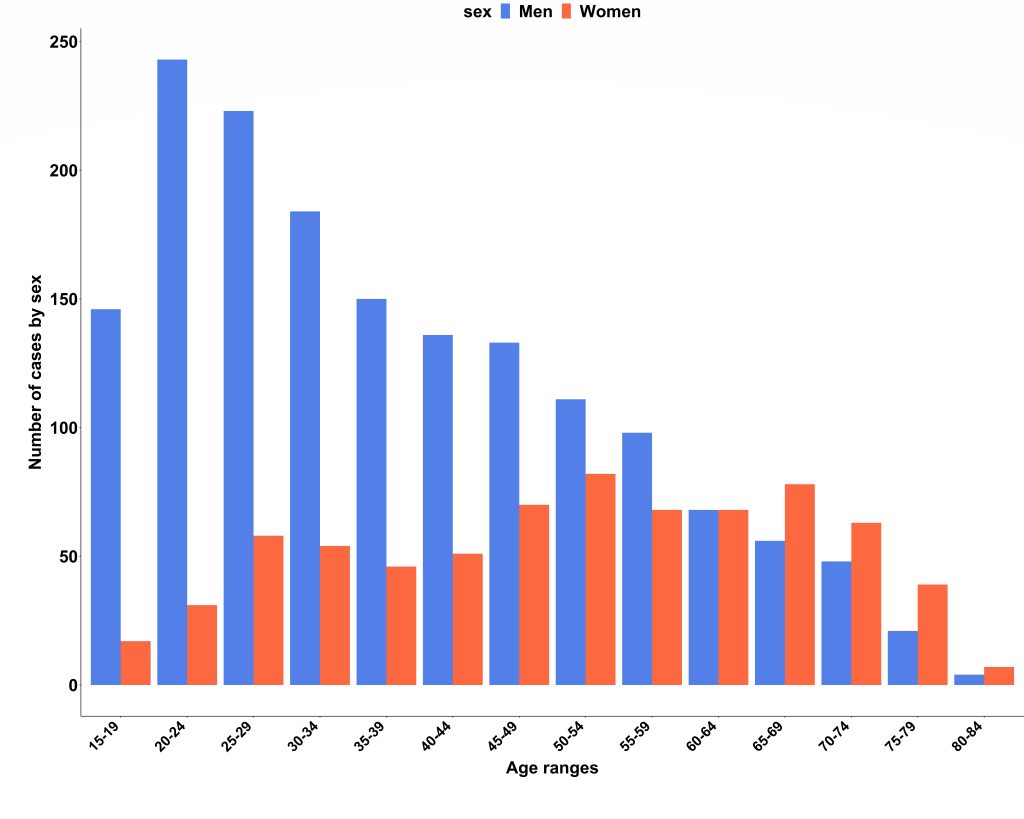
We obtained national hospital admission data from NHS Digital (3) and found there were 2353 admissions due to acute myocarditis over the 2-year study period (69% men; median age, 40 years; IQR, 27–55 years). Case ascertainment was calculated at 66%, and the London cohort was confirmed to be representative of national myocarditis admissions. Interestingly, we also found that men were significantly younger than women on admission to hospital (median age 35 years vs 52 years; P<0.001).
In conclusion, in this population-based study ~1 in 13 patients presenting with acute myocarditis were found to have an underlying variant in a gene robustly linked to DCM or ACM that would be reported as likely pathogenic in a patient with cardiomyopathy, compared with <1% in healthy controls. The presence of these variants affected clinical outcomes, particularly with DCM variants being associated with a trend towards greater all-cause mortality.
This study provides novel insights into why a small but important subset of patients with myocarditis experience major adverse events, whilst the majority usually recover spontaneously. It supports the concept that genotype-positive individuals may remain phenotypically silent until the occurrence of an environmental trigger.
This study suggests that genetic counselling and testing should be considered in patients with acute myocarditis, particularly in those with greater LV dysfunction, arrhythmia, or family history of cardiomyopathy. This may inform risk stratification and clinical management, including the need for ongoing surveillance and family screening, when cardiomyopathy-associated genetic variants are present.
A team of researchers with expertise across various disciplines was needed to harness the latest advances in precision phenotyping and genotyping whilst also leveraging national epidemiological datasets to deliver this study. Further work is underway to replicate these findings in a larger multi-centre setting and to provide greater mechanistic insights into genetic predisposition and disease progression in inflammatory cardiomyopathy using a systems biology approach.
The study was published under open access in Circulation on 26th Sept 2022:
Grun S, Schumm J, Greulich S, Wagner A, Schneider S, Bruder O, Kispert EM, Hill S, Ong P, Klingel K, et al. Long-term follow-up of biopsy-proven viral myocarditis: predictors of mortality and incomplete recovery. Journal of the American College of Cardiology. 2012;59:1604-1615
Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European heart journal. 2013;34:2636-2648, 2648a-2648d.
We are pleased to share our Letter to the Editor published in Genetics in Medicine (McGurk et al. 2022) in response to the latest American College of Medical Genetics & Genomics (ACMG) recommendations on the reporting of secondary findings from clinical sequencing data (ACMG SF) (Miller et al., 2021 a,b).
The increasing availability of genome sequencing in clinical practice provides opportunities for improvements in health. One specific opportunity is through the early detection of health risks that might allow for preventative intervention. Since 2015 the ACMG has recommended that whenever clinical genome sequencing is undertaken, a defined set of genes should be actively interrogated for DNA variants that might herald a preventable clinical risk, such as a strong inherited predisposition to a specific cancer.
This is a recommendation for a genetic screening programme. Before implementing screening, it is necessary to understand the benefits and harms of the screening test, and the benefits and harms of any downstream interventions triggered by a positive test. It is also important to understand the costs of the programme, and who will bear these costs.
Genes associated with inherited cardiac conditions, including cardiomyopathies (heart muscle disease), make up about half of the latest ACMG SF gene list. These conditions can lead to sudden cardiac death, and sadly a fatal heart rhythm problem is sometimes the first manifestation of disease. Identifying people with these conditions early, before they even develop symptoms, is the only way of preventing such deaths. This is the motivation for screening when genetic data become available through testing for an unrelated condition.
However, not everybody who carries a “disease-causing” variant in a cardiomyopathy gene will develop disease. We call this incomplete penetrance. And at present there is huge uncertainty around this – when someone in the population is found to have a “disease-causing” variant, we don’t really know what risk they have of developing cardiomyopathy, and/or of having a dangerous heart rhythm.
We have no doubt that some people will benefit from screening, but others will also be harmed, e.g. through psychological harms, a loss of time or money due to burdensome surveillance, and potentially genetic discrimination. We do not believe that we have sufficient understanding of the relative benefits and harms to recommend that this should be carried out as standard-of-care.
Patients should be advised of this uncertainty. If a patient chooses to be informed of secondary findings this should ideally be done in the context of research to understand the benefits and harms.
We were particularly motivated to address this following recent update to the list of genes recommended for screening. The ACMG have added TTN (titin) and FLNC (filamin C) to the list of cardiomyopathy genes to evaluate. We have been studying Titin for some years, and we think that the inclusion of this gene presents huge challenges. Variants that we would call “likely pathogenic” if found in someone with cardiomyopathy are actually found in about 1 in 200 people in the general population. The vast majority never develop disease. Is there a net benefit in looking for these variants? We used data from the UK biobank to estimate what the benefits of screening might be.
Secondary findings in FLNC
The authors of the new recommendations say that FLNC variants are highly penetrant, conferring a high risk of cardiomyopathy and arrhythmia. We agree that people with cardiomyopathy due to variants in FLNC are more likely have rhythm problems than people with some other forms of cardiomyopathy. However, it has not been shown that these variants are highly penetrant when found in people in the general population.
In this analysis we studied participants in the UK Biobank (UKBB) and found 50 individuals with rare heterozygous FLNC-truncating variants (a similar prevalence to that seen in consortium populations) that would meet criteria for a P/LP annotation and might be returned as secondary findings. None of these individuals were apparently diagnosed with cardiomyopathy. The benefits of screening for these variants are unclear.
Secondary findings in TTN
0.44% of UKBB participants carried rare (heterozygous) TTN-truncating variants, TTNtv. 1.4% of these were known to have cardiomyopathy when they signed up for the biobank study. These are therefore not strictly secondary findings.
A further 2.4% (1 in 40) showed evidence of a previously unrecognised cardiomyopathy on a one-off cardiac imaging assessment (derived from the proportion of heterozygous who met criteria for DCM during the UKBB cardiac MRI – CMR). This gives us an estimate of the number of people we would diagnose with cardiomyopathy if we offered people with SFs in TTN a one-off assessment.
Further individuals developed cardiomyopathy over time. To detect these would require ongoing surveillance, likely with CMR repeated at intervals. We estimate that this would detect a further ~3 incident cases per 1,000 person-years of ongoing surveillance in the population of people found to have TTN variants as secondary findings (200 people under surveillance for 5 years each = 1000 person-years).
Modelling adverse outcomes
So far, we have looked at how many people will develop cardiomyopathy. What we are really interested in is how many people will run into problems that might be preventable by early detection and intervention, particular sudden death.
Based on previous estimates that about 1 in 25 people with established dilated cardiomyopathy might die suddenly over a four-year window, we estimated that to prevent one death we would need to make 25 new diagnoses of cardiomyopathy, which would require us to carry out ~8,000 person-years of surveillance in people found to have TTN variants as secondary findings.
In other words, if we called back 1600 people who had a normal CMR, and repeated the test 5 years later, we might find 25 people who had now developed DCM, and treating these might prevent a single sudden death.
This assumes that the death is entirely preventable with early diagnosis and treatment, which is likely over-optimistic. Moreover, it ignores the fact that some of these people will not benefit from screening because they have other more immediate health concerns – e.g., they may have had genetic testing because of a cancer, or another rare genetic condition.
An alternative methodology (based on the total observed excess mortality amongst TTN variant carriers, and optimistically assuming that this excess mortality is entirely preventable) suggests that we would need to enrol 100 people into long-term surveillance to prevent 1 death in 10 years.
We therefore believe that while some people will benefit from return of secondary findings, it is premature to recommend this as standard-of-care until we have a better understanding of the benefits and harms of such screening.
Response to McGurk et al.
The authors of the ACMG recommendations responded to our letter (Gollob et al. 2022). They pointed out that the UKBB cohort may underestimate disease prevalence and incidence. We acknowledge the likelihood of survivorship bias in the UKB in our letter but note that the prevalence of DCM is very close to other population estimates, so do not believe that this bias has distorted our estimates importantly.
Furthermore, the UKB cohort likely provides very reasonable estimates for opportunistic screening carried out in adults, e.g., for adult cancer patients, or healthy adults undergoing clinical sequencing because they have a child with a rare genetic condition (for trio analyses).
The authors agreed that more studies are needed on the costs and benefits of reporting SFs and add that they can adapt the future gene lists as new evidence becomes available.
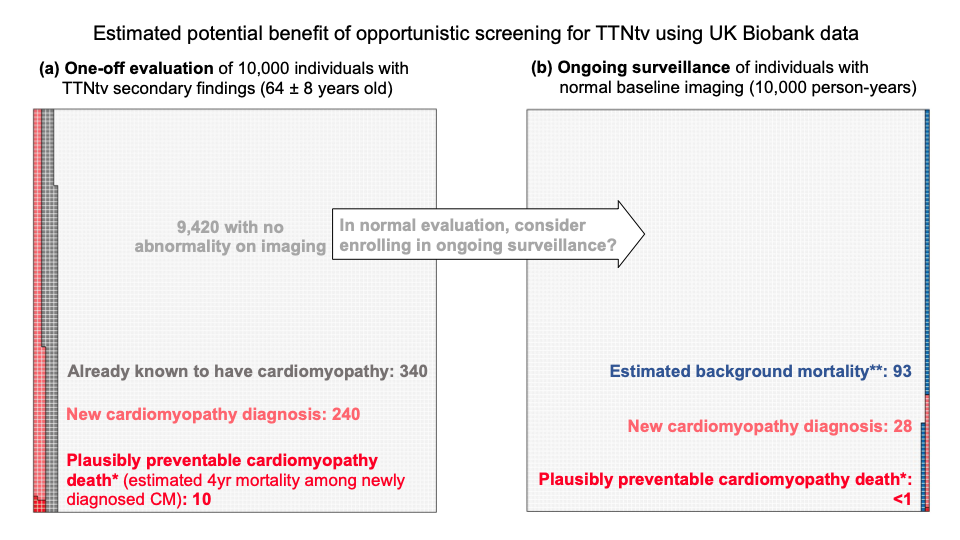
Figure – Visual representation of the potential benefits of opportunistic screening for TTNtv estimated using UK Biobank data. A) Expected findings for a one-off cardiac imaging evaluation of 10,000 individuals with a TTNtv identified as a secondary finding. We estimate that 340 individuals would already be known to have cardiomyopathy, and 240 would be newly identified. CM-related mortality has been estimated as ~4%/4years11,12, so we expect ~10 deaths in this timeframe amongst the newly identified cases, which might plausibly be preventable. This is one estimate of the benefit of opportunity screening, though it is not yet known to what extent early identification would prevent these, particularly given that this population will have other competing risks. Since they are undergoing clinical sequencing for another indication, the morbidity and mortality of that condition would influence the overall value of screening for secondary findings. B) For the 9,420 individuals with no abnormality on initial imaging, we need to consider the potential value of ongoing surveillance, e.g., with serial imaging. For each 10,000 person-years of surveillance we expect 28 new cardiomyopathy diagnoses, with <1 plausibly preventable cardiomyopathy-related death in the next four years (based on mortality rates described above). For comparison, the estimated background mortality in the UK is 93 in 10,000 person-years. The background mortality in a population undergoing clinical sequencing for another indication may be higher. These estimates are based on the UK biobank population, a population of older adults around the typical age of presentation for dilated cardiomyopathy. The yields of one-off and serial evaluation might be expected to be lower in younger individuals. The potential benefit of opportunistic screening for sudden cardiac arrest prevention can also be estimated by directly measuring the incidence of sudden cardiac arrest. Amongst TTNtv heterozygotes in the UK biobank (including individuals already known to have disease, as well as those newly recognised) the incidence is 5/10,132 person-years, i.e., 0.05%, compared with a background SCA incidence of 0.04% in the remainder of the cohort (P>0.05). *Plausibly preventable cardiomyopathy mortality was estimated as 4%/4yr among newly diagnosed cardiomyopathy11,12. **mean death rate in 2019 of registered deaths in 59-69-year-olds in the UK by the Office for National Statistics. The data supporting this figure can be found in the supplementary methods, Table S1, and Figure S1. Of note, these estimates do not account for competing risks.
Researchers from the Cardiovascular Genetics & Genomics Group have published their latest findings in Open Heart journal, using the Heart Hive platform to connect with people with cardiomyopathy. The paper assesses the effects of the first wave of COVID-19 pandemic on patients with cardiomyopathy using three parallel approaches.
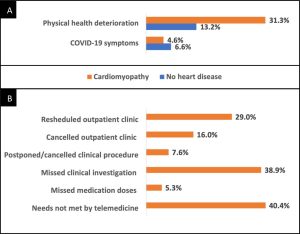
The study has demonstrated that a third of the surveyed patients with cardiomyopathy felt that their physical health deteriorated as a result of the pandemic when assessed shortly after the first wave of the pandemic. This was a significantly higher proportion than those that had reported a suspected COVID-19 infection.
Reported changes in physical health for patients with cardiomyopathy and subjects without heart disease in the Heart Hive COVID-19 study demonstrating that significantly more patients with cardiomyopathy experienced subjective deterioration in physical health. (A) and the reported provision of healthcare for patients with cardiomyopathy during the pandemic (B).
The study further illustrated that many patients had had outpatient clinic appointments rescheduled (29%) or cancelled (16%), and missed clinical investigations (38.9%), procedures (7.6%) or doses of medication (5.3%). Additionally, 40.4% of patients with cardiomyopathy felt that their health needs could not be met by telemedicine.
The findings also indicated that the psychological impact of the pandemic did not differ significantly between patients with cardiomyopathy and subjects without heart disease. Patients with cardiomyopathy did feel that they may be more susceptible to COVID-19 infection and suffer more severe illness if infected.
Approximately 1 in 5 patients with cardiomyopathy had received a recommendation to ‘shield’; a higher proportion of the patients that had received a recommendation to shield reported worsening of their cardiomyopathy symptoms.
We also conducted two other parallel research initiatives alongside the Heart Hive COVID-19 study, which are also published in this article. By studying participants of the Royal Brompton & Harefield Hospital Cardiovascular Research Centre Biobank, we found that patients with DCM and HCM were no more likely to be infected with COVID-19 than the rest of the UK population. However, of the patients with DCM and HCM that were studied, those that had been infected with COVID-19 had more frequently required treatment in hospital during the first wave of the pandemic.
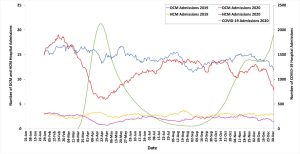
Finally, we used NHS Digital Hospital Episodes Statistics to illustrate that there was a 17.9% year on year reduction in cardiomyopathy-related hospital admissions in 2020, with the lowest hospitalisation rate occurring during the first UK lockdown.
The impact of the COVID-19 pandemic on hospital admissions with a primary diagnosis of dilated cardiomyopathy or hypertrophic cardiomyopathy across NHS England, 2019–2020. Hospital admission data are presented as 28-day moving averages. DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; NHS, National Health Service.
To the best of our knowledge, this is the first study to assess both the direct and indirect effects of the COVID-19 pandemic on patients with cardiomyopathy. We hope these findings can be used to adapt clinical services to meet patients’ health needs as the pandemic evolves.
We are very grateful to the Heart Hive participants who contributed to this research initiative and to Cardiomyopathy UK for their ongoing support of this study and the Heart Hive. If you’re taking part in the study, please keep completing your COVID surveys to help us see how COVID-19s effect on people with cardiomyopathy has been changing during the course of the pandemic and with the developments of vaccines and new treatments.
If you haven’t signed up the the Heart Hive and would like to, you can find out more on the Heart Hive home page.
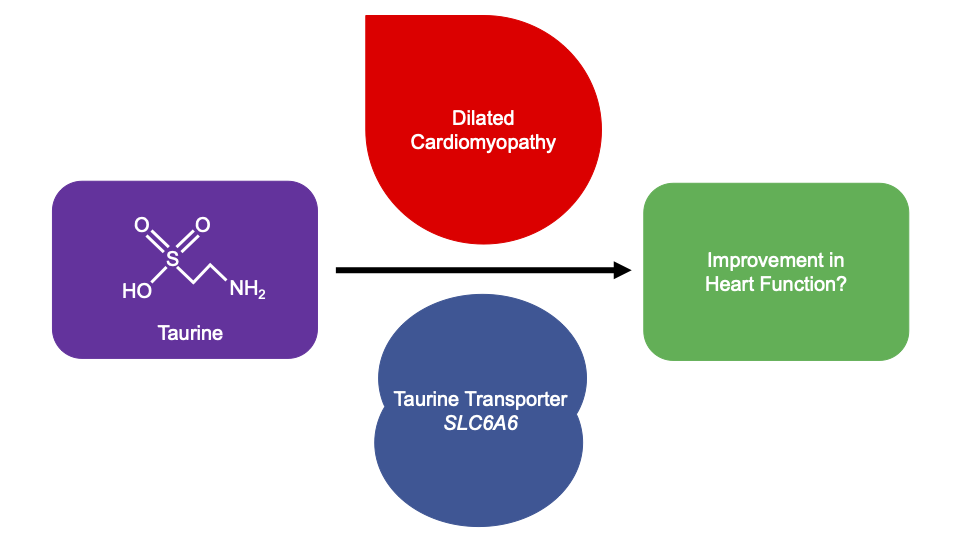
The Cardiovascular Genetics and Genomics group is delighted to share the systematic review in Wellcome Open Research (McGurk et al. 2022) entitled “Effect of taurine administration on symptoms, severity, or clinical outcome of dilated cardiomyopathy and heart failure in humans: a systematic review”.
Taurine, 2-aminoethanesulfonic acid, is an essential amino acid found in animal products. Taurine is produced for human consumption as a supplement and ingredient in beverages. Supplementation is a safe, inexpensive, and effective treatment for dilated cardiomyopathy (DCM) in domestic mammals (Pion et al. 1987; Pion et al. 1992; Moise et al. 1991; Keith et al. 2001; Kittleson et al. 1997), however it is currently unlicensed in Europe and the United States for human medical treatment. Recent genome-wide association studies of DCM have identified the locus of the taurine transporter (SLC6A6) (Garnier et al. 2021; Tadros et al. 2021). Additionally, a variant in SLC6A6 has been identified in a consanguineous family with retinal degeneration and mild hypokinetic cardiomyopathy with systolic dysfunction and systolic dilatation of the left ventricle, which corrected with taurine supplementation after 24-months (Ansar et al. 2020). It is unknown whether taurine supplementation can improve human DCM, and furthermore whether improvements are observed when taurine levels are in normal reference ranges.
To assess whether taurine supplementation may be a novel therapeutic option for DCM, we undertook a systematic review. Four electronic databases were searched until 11/03/21. 285 articles were identified, of which eleven met our criteria for inclusion. Taurine supplementation varied across studies; by dose (500 mg to 6g per day), frequency (once to thrice daily), delivery method (tablet, capsule, drink, powder), and duration (2 to 48 weeks). Patient inclusion was all-cause HF patients with ejection fraction (EF) <50% and no study was specific to DCM. While improvements in diastolic and systolic function, exercise capacity, and haemodynamic parameters were described, only EF and stroke volume were measured in enough studies to complete a meta-analysis; the association was not significant with all-cause HF (P<0.05). No significant safety concerns were reported. A formal clinical trial is needed to address whether taurine supplementation is beneficial to the approximately 1/250 individuals with DCM in the population.
How this research came about
The decrease in charity research funding was well publicised in 2020 due to the coronavirus pandemic. I began trying to identify alternative funding sources for future independent research funding. At the time I stayed across the road from a Cats Protection shop. With plenty of time on my hands after work due to UK lockdowns and being new to the field of cardiomyopathy research (without the limits of knowing what is already known), I decided to google whether cats get inherited cardiomyopathies.
Taurine is a regular supplementation for domestic cats presenting with DCM as they lack the ability to create taurine (there are theories around the lack of access of domestic cats to taurine-rich mice). Further research showed that this treatment has been replicated in other domesticated animals. As a geneticist, I did a brief analysis of the taurine metabolic pathway to identify genes where variants may have a potential role in bodily taurine variation, and then assessed the results from a DCM GWAS study we recently published, and I noticed that that one of the hits was in the locus of the taurine transporter (nearest gene LSM3). The lead SNPs are upstream to, and confirmed thyroid eQTLs on GTEx of, the taurine transporter (SLC6A6 or TauT). Without going into detail – the more I dug, the more I found that implicated taurine in DCM and/or altered heart phenotypes.
Why hasn’t a clinical trial for DCM in humans been undertaken?
taurine tablets are cheap and available (no financial gain?)
human plasma measurements may be increased by monocyte activation and thus may be inaccurate.
the mechanism of taurine action is mostly unknown.
DCM GWAS with large sample sizes have only been published very recently.
There is a lack of intersection between veterinary medicine and human sciences (diet-associated DCM in dogs has a group on facebook with 114k members…).
Big thank you to the many members of the group for their support – Rachel Buchan and Katherine Josephs provided much needed sanity checks, James Ware supported the idea from the beginning, Melpi Kasapi helped with the systemmatic review, as well as Paul Barton, Stuart Cook, Brian Halliday, Declan O’Regan, Sean Zheng, Ang Roberts, for discussions.
Ansar M, Ranza E, Shetty M, et al.: Taurine treatment of retinal degeneration and cardiomyopathy in a consanguineous family with SLC6A6 taurine transporter deficiency. Hum Mol Genet. 2020; 29(4): 618–623.
Garnier S, Harakalova M, Weiss S, et al.: Genome-wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur Heart J.; 2021; 42(20): 2000-2011.
Keith ME, Ball A, Jeejeebhoy KN, et al.: Conditioned nutritional deficiencies in the cardiomyopathic hamster heart. Can J Cardiol. 2001; 17(4): 449–458.
Kittleson MD, Keene B, Pion PD, et al.: Results of the multicenter spaniel trial (MUST): taurine- and carnitine-responsive dilated cardiomyopathy in American cocker spaniels with decreased plasma taurine concentration. J Vet Intern Med. 1997; 11(4): 204–211.
McGurk KA, Kasapi M and Ware JS. Effect of taurine administration on symptoms, severity, or clinical outcome of dilated cardiomyopathy and heart failure in humans: a systematic review. Wellcome Open Res 2022, 7:9.
Moise NS, Pacioretty LM, Kallfelz FA, et al.: Dietary taurine deficiency and dilated cardiomyopathy in the fox. Am Heart J. 1991; 121(2 Pt 1): 541–547.
Pion PD, Kittleson MD, Rogers QR, et al.: Myocardial failure in cats associated with low plasma taurine: A reversible cardiomyopathy. Science. 1987; 237(4816): 764–768.
Pion PD, Kittleson MD, Thomas WP, et al.: Response of cats with dilated cardiomyopathy to taurine supplementation. J Am Vet Med Assoc. 1992; 201(2): 275–284.
Tadros R, Francis C, Xu X, et al.: Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021; 53(2): 128-134.
The Cardiovascular Genetics and Genomics group is delighted to share the results of a new, multi-centre study on left-ventricular noncompaction (LVNC), published in Genetics in Medicine here.
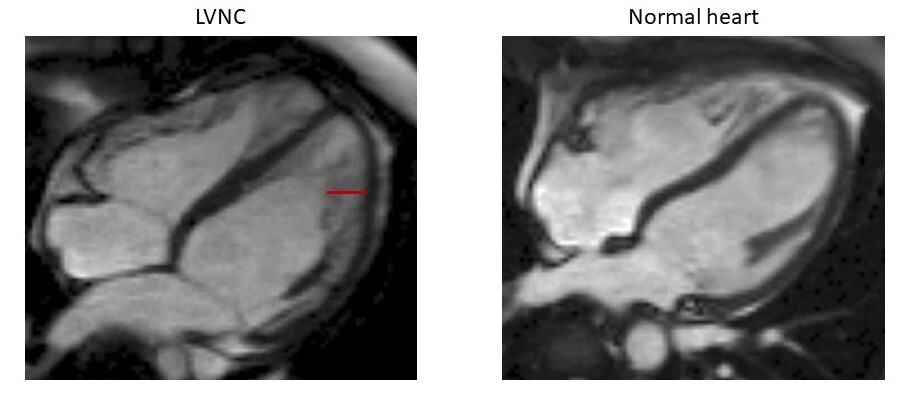
LVNC is characterized by a partially noncompacted heart muscle, with a classically “spongy” appearance at imaging and diagnostic criteria based on the relative amount of compacted and noncompacted myocardium, as displayed in the figure below. Such diagnostic criteria though — when applied using the newer imaging technologies — can lead to the detection of LVNC in up to 15% of the population, highlighting a high risk of over-diagnosis.
Hearts in 4-chamber view of a patient affected with LVNC (left) and a healthy individual (right), obtained with cardiac magnetic resonance imaging. The non-compacted portion of the myocardial layer in the patient with LVNC is highlighted with a red line.
The true nature of LVNC as a clinical entity is subject to much debate as to LVNC represents a separate disease or whether it is to be considered a secondary trait that can manifest in presence of an underlying cardiomyopathy. Such discussion is still far from reaching a definitive answer, as reflected by the two different classifications assigned by the European Society of Cardiology and the American Heart Association, which label LVNC as an “unclassified cardiomyopathy” and a “primary genetic cardiomyopathy”, respectively.
In this study, developed in collaboration with Amsterdam UMC, the University of Florence, the Aswan Heart Centre and the Partners’ Healthcare Laboratory for Molecular Medicine, we have:
collected genetic data from over 800 patients referred for genetic testing for LVNC with LVNC (575 from previously published cohorts and 264 unpublished) and compared the frequencies of rare (carried by fewer than 1 in 10000 individuals) coding variants in 70 genes with those observed in a publicly available reference population (Genome Aggregation Database [gnomAD], ~125,000 individuals): this comparison showed which genetic variant classes are significantly associated with LVNC.
compared results with those obtained utilizing the same strategy on cohorts of patients with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), in previous studies performed by our group: this analysis’ main purpose was to assess the genetic overlap between LVNC and these two other cardiomyopathies, so to estimate the proportion of cases in which non-compaction may be a secondary trait of underlying DCM or HCM, rather than a distinct process.
Analyzed cardiac phenotypes of population individuals carrying specific LVNC-associated variants, so to assess the effect of such variants in the population and investigate whether their presence correlates with sub-clinical phenotypes: this genotype-phenotype analysis serves to understand if genetics can help discern between physiological and pathological noncompaction.
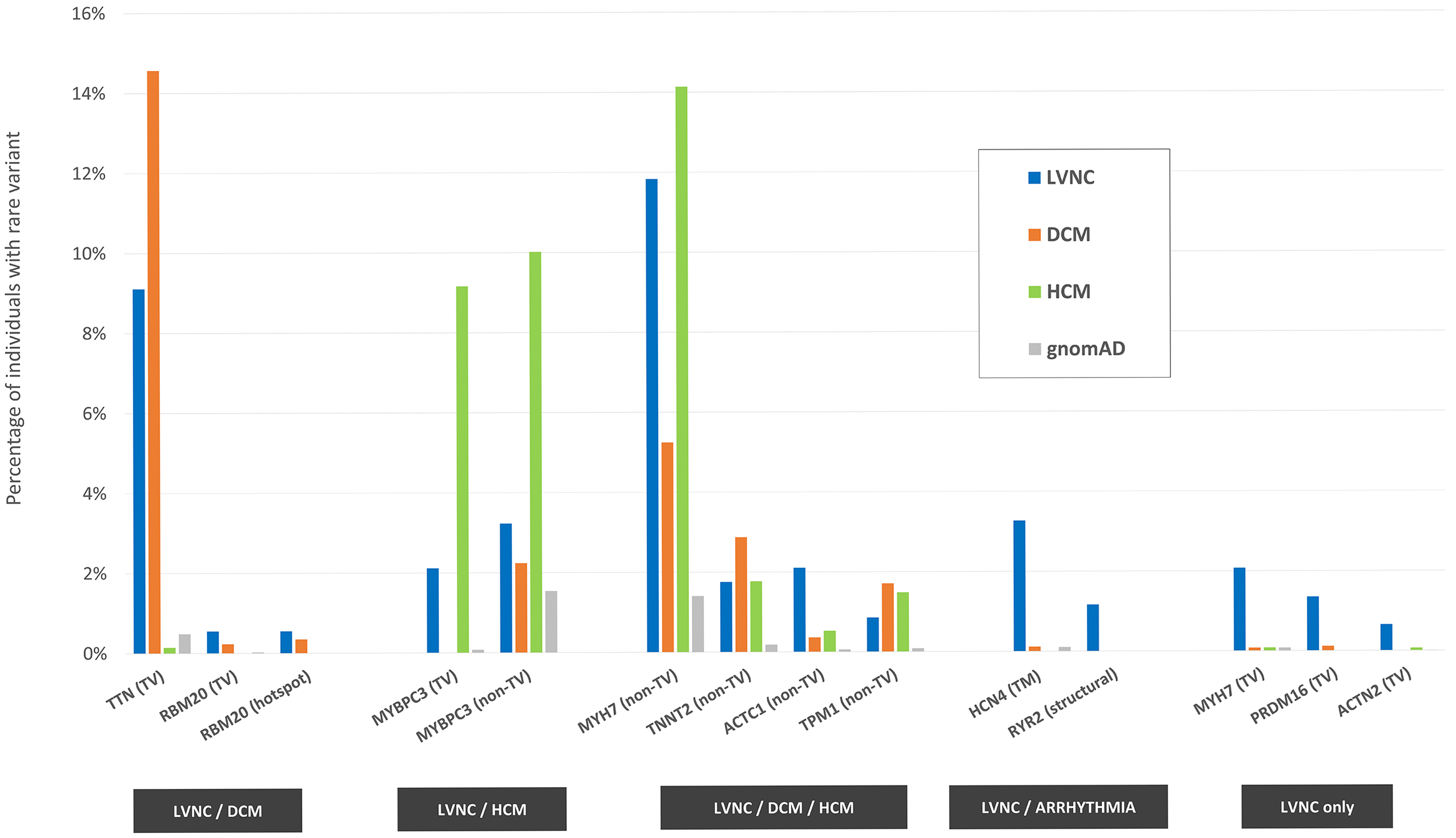
One figure from the paper, recapitulating the main results: 14 variant classes (in 11 genes) are associated with LVNC, having been found significantly more often in LVNC patients compared with population individuals (% indicated by the blue bars vs % indicated by the gray bars). Such variant classes are estimated to explain approximately 37% of cases. The disease association for the majority of them (11, in 9 genes) is shared with DCM (first group from the left, orange bars), HCM (second group, green bars) or both (third group). The associations with variants in the HCN4 and RYR2 genes are shared between LVNC and arrhythmogenic conditions, while the associations with truncating variants in MYH7, PRDM16 and ACTN2 are unique to LVNC. TV=truncating variants, TM=transmembrane, hotspot=residues 634-638 in RBM20.
As shown in the figure, the vast majority of LVNC-associated variant classes are also causative of HCM and/or DCM, suggesting how in a majority of cases LVNC is probably to be considered a secondary trait manifesting in patients affected with an underlying primary cardiomyopathy. However, there are certain cases (estimated in approximately 1 in 20 in the analyzed cohorts) in which LVNC is observed in presence of genetic variants that are uniquely LVNC-associated, suggesting how LVNC can also present as an isolated, distinct disease entity.
The association shared between LVNC and arrhythmogenic conditions with variants in the ion channel genes HCN4 and RYR2 (associated with sinus node disease and catecholaminergic polymorphic ventricular tachycardia, respectively) highlights how a subset of LVNC patients may be at risk of life-threatening arrhythmogenic events. Systematic screening for variants in these genes in LVNC patients may aid the early identification of such cases.
Of the variant classes associated uniquely with LVNC, truncating variants in MYH7 are of particular interest, as heterozygous “loss-of-function” variants have not previously been associated with cardiomyopathies (in contrast with non-truncating variants in the same gene, which are among the most important genetic causes of HCM and DCM). In this study, besides the significant association of these variants with LVNC, we also found how individuals in the general population (including the UK Biobank) carrying this type of variants have a significantly higher degree of ventricular noncompaction compared with non-carriers.
Taken together, these results contribute to the characterization of LVNC and suggest how:
in the majority of cases – LVNC is a morphological phenotype manifesting in presence of an underlying cardiomyopathy.
in approximately 5% of the cases, LVNC appears to be caused by a distinct genetic aetiology which suggests it can also be a distinct disease entity.
in addition, results obtained on phenotyped population cohorts confirm a concrete danger of LVNC over-diagnosis, suggesting how diagnostic criteria for LVNC should not be based solely on imaging, but conjugated e.g. with genetic screening to help identify individuals at risk of developing left-ventricular dysfunction.
We are delighted that the flagship publications describing the Genome Aggregation database (nomAD) have appeared today in Nature, Nature Medicine, and Nature Communications.Members of the CV Genetics & Genomics team have been long-term contributors to the gnomAD resource (and its predecessor, the exome aggregation consortium ExAC), and we have hugely enjoyed being part of this collaboration.Today’s package of publications includes two papers led by members of our team. “Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals” has already been described in a previous blog post when we made our paper open access on the bioRxiv preprint server a year ago (see here), and leverages the power of gnomAD to annotate a particular group of variants that can cause severe rare genetic diseases. Rather than disrupting the sequence of the gene itself, these variants lie just upstream of the gene and interfere with regulatory signals that control translation of genes into proteins. This study was a home-grown effort, spear-headed by Nicky Whiffin while working at Imperial. The second study was also co-led by Nicky, and arose from a visit to spend some time working with Daniel MacArthur’s team at the Broad Institute. “The effect of LRRK2 loss-of-function variants in humans looks at a specific gene, LRRK2, suggested as a therapeutic target for Parkinson’s disease. There have been some concerns about the safety of targeting LRRK2 with drugs. However, Nicky and colleagues, working with data from gnomAD, 23andMe, and UK Biobank, found that individuals who carried genetic variants that inactivate one copy of this gene did not have any apparent adverse consequences. This provides reassurance that therapeutic inhibition of LRRK2 is likely to be well-tolerated and safe.You can read Nicky’s reflections on her work and the gnomAD package on her blog on her new webpage, which she has recently launched as she prepares to move to Oxford to set up a new lab at the Wellcome Centre for Human Genetics.
The years preceding the availability of large, publicly accessible cohorts of genetically characterized individuals witnessed a large number of studies implicating genes in diseases such as dilated cardiomyopathy (DCM), without sufficiently robust evidence according to today’s standards.
Projects such as the 1000 Genome Project, the Exome Sequencing Project and the Exome Aggregation Consortium (ExAC) database showed how, collectively, rare variants were considerably more common than had been expected in the population, implying that rarity is necessary but not sufficient to deem variants pathogenic under a dominant monogenic model assuming high penetrance, and casting doubts on many previously proposed gene-disease associations.
Here, we re-evaluate 56 genes in which rare variants have previously been reported to be associated with DCM. We have gathered data from more than 2,500 patients with DCM, and compare genetic variation in these individuals with control populations to provide a much needed re-assessment of the genetic architecture of monogenic DCM.
In summary:
– We analysed rare (carried by fewer than 1/10,000 individuals) genetic variants in 56 genes previously implicated in DCM.
– We first compared genetic data from two primary cohorts of ~1,000 DCM cases and ~1,000 healthy controls (with normal cardiac MRI scans) uniformly sequenced and processed thorough identical pipelines, to minimize technical bias.
– We also analysed a secondary cohort of ~1,500 DCM cases to encompass different patient profiles, and compared them to a population reference cohort (~60,000 ExAC individuals) as an additional population control cohort to maximise statistical power.
– ExAC is not a control cohort as such, and comprises whole-exome sequencing data. These two key characteristics, representing potential sources of bias, were addressed applying several quality control steps to minimize artefactual stratification.
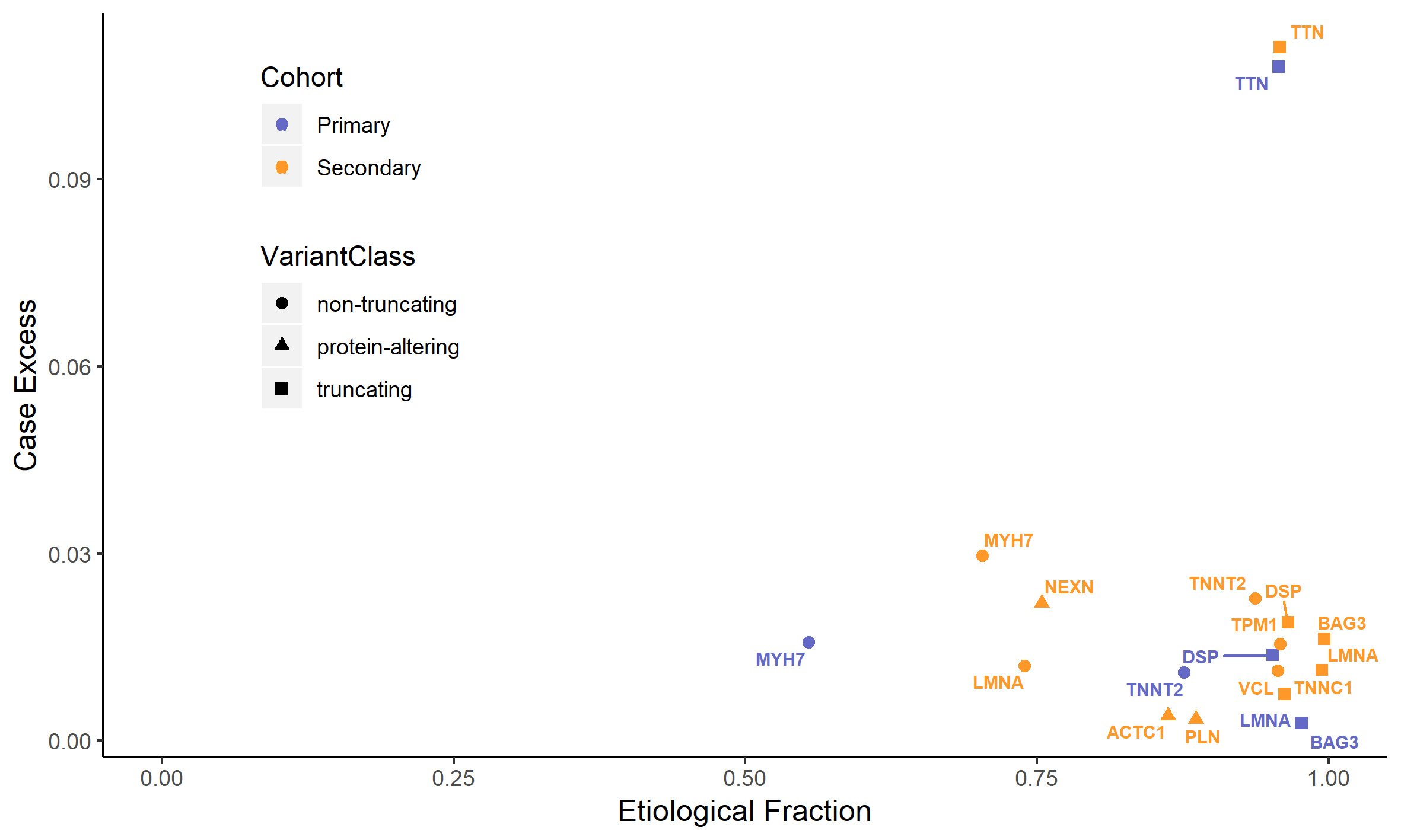
– Across three separate comparisons, we detected robust statistical evidence for association with DCM for only 12 of the 56 genes (TTN, MYH7, LMNA, TNNT2, DSP, BAG3, TPM1, TNNC1, VCL, NEXN, ACTC1 and PLN).
– We computed the corresponding Etiological Fraction for each of the DCM-associated variant classes, which is an estimate of the probability that a variant (of the same class) is the cause of disease when it is found in a patient. Some variant classes, including all those predicted to result in a truncated protein, have Etiological Fraction values >0.95, indicating very high probabilities of pathogenicity even before evaluating additional evidence.
– Specific variant classes in VCL and TPM1 were observed significantly more often than expected in patients younger than 18 years of age, suggesting that these variants may primarily cause early-onset DCM.
One of the figures from the paper: the vertical axis displays an estimate of what proportion of DCM cases is due to variants in each gene. As an example, truncating variants in TTN explain approximately 10% of DCM. The horizontal axis displays the prior probability that a variant detected in a patient is the cause of disease, which corresponds to a measure of how interpretable variants are in the diagnostic setting.
These results do not rule out the other 44 analysed genes from playing a role in DCM, but they suggest that their contribution will be negligible or confined to very specific variant classes. This data will be instrumental to DCM genetic testing, both in the composition of diagnostic gene panels and in the interpretation of variants detected in patients, and to community gene curation efforts such as the one carried out by the ClinGen consortium, evaluating also other lines of evidence.
We are excited to share the recent press release and BBC coverage of our study into genetic factors contributing to cancer drug related cardiomyopathy.
In this study, published in the journal Circulation, we analysed genes of more than 200 cancer patients from the UK, Spain and the US who had been diagnosed with a type of heart condition called cancer-therapy induced cardiomyopathy, or CCM. We found patients who developed the heart condition were more likely to carry genetic faults linked to cardiomyopathy – and in particular that patients were more likely to carry a faulty version of a gene called titin.
We are excited to share our work using gnomAD to characterise 5’UTR variants that create or disrupt upstream open reading frames (uORFs) and explore their role in disease: https://www.biorxiv.org/content/10.1101/543504v2
We show that these variants are under strong negative selection (indicative of being deleterious), and identify a subset (that form ORFs overlapping the coding sequence) with signals of selection equivalent to coding missense variants.
We find increased signals of selection when these variants occur in the 5’UTRs of curated haploinsufficient, LoF intolerant and known dominant LoF developmental disease genes, supporting a loss-of-function effect of these variants on translation.
We identify specific genes where uORF perturbation appears to be an important disease mechanism (e.g. NF1 and IRF6), and report a novel uORF frameshift variant in NF2 that segregates with disease in two families with neurofibromatosis.
Our approach illustrates the power of using large population databases and grouping non-coding bases by functional effect, to identify subsets of variants that are highly deleterious. Although the strength of selection at the level of UTRs is equivalent to synonymous variants, we see a much stronger signal at these specific uORF-perturbing sites.
In our previous posts (June 2017 and October 2017) we explained two of our studies looking at the role of truncating variants in the Titin gene (TTNtv) in dilated cardiomyopathy (DCM). In the first of these, we found that having a TTNtv meant that a DCM patient was more likely to have had a past history of abnormal heart rhythms when they were first diagnosed with DCM. But we did not have enough data to definitively assess whether this was due to rhythms in the upper (atrial) chamber of the heart, the bottom (ventricular) chamber, or both. We also didn’t know whether this meant that DCM patients with a TTNtv were at higher long-term risk of potentially dangerous or life-threatening heart rhythm abnormalities. In our second study, we followed up a large group of patients with DCM but didn’t find an increased rate of major arrhythmic events in patients with a TTNtv. As a whole this group had relatively mild symptoms of DCM at the time of the study (the majority being in NHYA class I/II heart failure), with moderately impaired ventricular function – which puts them in a comparatively low-risk group for arrhythmias. Perhaps as a result, the overall rate of arrhythmic events in the study was low, making it harder to see subtle differences between those with and without a TTNtv.
To investigate further, we therefore started a new study on patients with DCM who also had an implantable cardiac device (ICD or CRTD). Not only are patients with these devices more likely to have an arrhythmia (they are typically given to the patients because they have worse heart function or symptoms) but, crucially, they monitor the patient’s heart rhythm 24-hours a day, 7-days a week. This allowed us to get complete coverage of each patient’s heart rhythm data over a number of years.
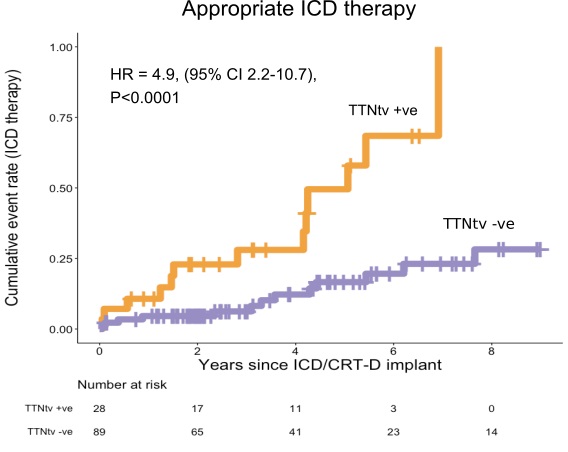
Our study, which was published earlier this month in JAMA: Network Open, found that TTNtv are associated with a significantly higher risk of both ventricular and atrial arrhythmia in DCM patients with an ICD or CRTD.
We had a strict definition of arrhythmia, for example only counting a ventricular arrhythmia if it was both fast (>200 beats per minute) and prolonged. These are the rhythms considered to pose the most danger to patients.
Importantly, we looked at the other major factors known to increase the risk of arrhythmia – age, male sex, level of left heart dysfunction and presence of heart fibrosis (scarring) on MRI – and even after controlling for these statistically TTNtvs were a strong predictor of arrhythmia.
Heart fibrosis, or scarring, on MRI is worth delving into a little more deeply. Fibrosis shows up as white patches on the heart in an MRI scan after a contrast agent (gadolinium) has been given via a vein. The presence of this fibrosis has long been known to be a strong predictor of arrhythmia, it’s likely that the scarred area interferes with the way electrical signals travel through the heart. DCM patients with a TTNtv don’t seem to have more fibrosis than other DCM patients – we found that in a previous study and confirmed it again here. However, a remarkable finding in our study was the apparent additive nature between fibrosis and a TTNtv in terms of arrhythmia risk. Of those patients that had both fibrosis and a TTNtv, 62% experienced a ventricular arrhythmia, compared to only 5% of those with neither fibrosis or a TTNTv.
So how might TTNtvs lead to more arrhythmia in DCM?
This study wasn’t designed to answer this question and much more work needs to be done. But one avenue worth investigating is the fact that DCM patients with a TTNtv seem to have thinner heart muscle walls than those without a TTNtv, as we discussed in one of our previous blogs from October 2017. Thinner walls lead to increased ‘strain’ on the heart muscle cells, which can potentially alter their electrical properties.
Can knowing if a patient has a TTNtv help decide whether or not they should have an ICD? This was a relatively small study and the robustness of the findings certainly needs to be tested in much larger cohorts. Such studies are under way (see our blog post from October 25th 2018) and it is hoped that in the future TTNtv may be one of several factors that can help in risk-stratifying patients with DCM.