The years preceding the availability of large, publicly accessible cohorts of genetically characterized individuals witnessed a large number of studies implicating genes in diseases such as dilated cardiomyopathy (DCM), without sufficiently robust evidence according to today’s standards.
Projects such as the 1000 Genome Project, the Exome Sequencing Project and the Exome Aggregation Consortium (ExAC) database showed how, collectively, rare variants were considerably more common than had been expected in the population, implying that rarity is necessary but not sufficient to deem variants pathogenic under a dominant monogenic model assuming high penetrance, and casting doubts on many previously proposed gene-disease associations.
Here, we re-evaluate 56 genes in which rare variants have previously been reported to be associated with DCM. We have gathered data from more than 2,500 patients with DCM, and compare genetic variation in these individuals with control populations to provide a much needed re-assessment of the genetic architecture of monogenic DCM.
The study has been published two days ago by the journal Circulation
(https://www.ahajournals.org/doi/abs/10.1161/CIRCULATIONAHA.119.037661).
In summary:
– We analysed rare (carried by fewer than 1/10,000 individuals) genetic variants in 56 genes previously implicated in DCM.
– We first compared genetic data from two primary cohorts of ~1,000 DCM cases and ~1,000 healthy controls (with normal cardiac MRI scans) uniformly sequenced and processed thorough identical pipelines, to minimize technical bias.
– We also analysed a secondary cohort of ~1,500 DCM cases to encompass different patient profiles, and compared them to a population reference cohort (~60,000 ExAC individuals) as an additional population control cohort to maximise statistical power.
– ExAC is not a control cohort as such, and comprises whole-exome sequencing data. These two key characteristics, representing potential sources of bias, were addressed applying several quality control steps to minimize artefactual stratification.
– Across three separate comparisons, we detected robust statistical evidence for association with DCM for only 12 of the 56 genes (TTN, MYH7, LMNA, TNNT2, DSP, BAG3, TPM1, TNNC1, VCL, NEXN, ACTC1 and PLN).
– We computed the corresponding Etiological Fraction for each of the DCM-associated variant classes, which is an estimate of the probability that a variant (of the same class) is the cause of disease when it is found in a patient. Some variant classes, including all those predicted to result in a truncated protein, have Etiological Fraction values >0.95, indicating very high probabilities of pathogenicity even before evaluating additional evidence.
– Specific variant classes in VCL and TPM1 were observed significantly more often than expected in patients younger than 18 years of age, suggesting that these variants may primarily cause early-onset DCM.
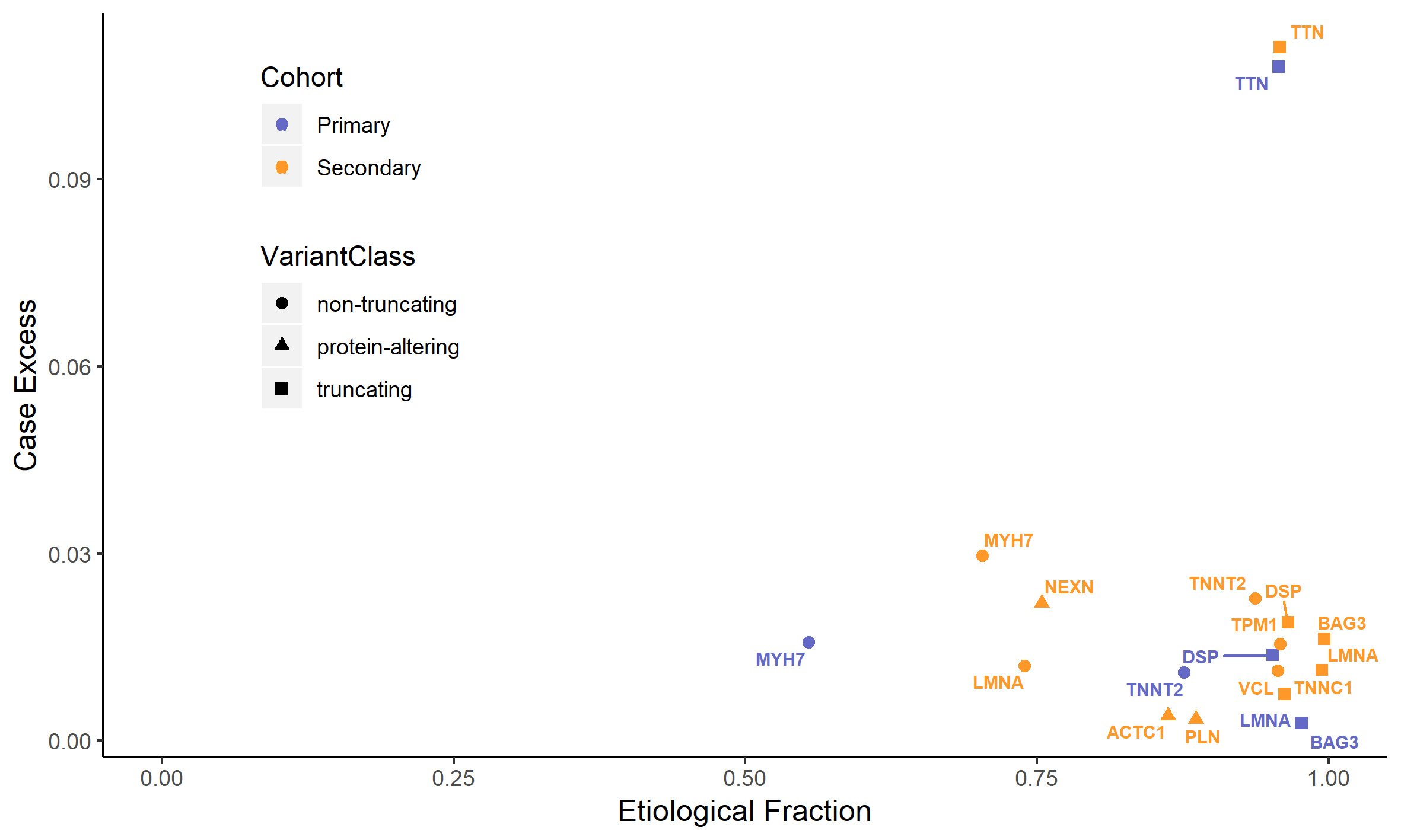
One of the figures from the paper: the vertical axis displays an estimate of what proportion of DCM cases is due to variants in each gene. As an example, truncating variants in TTN explain approximately 10% of DCM. The horizontal axis displays the prior probability that a variant detected in a patient is the cause of disease, which corresponds to a measure of how interpretable variants are in the diagnostic setting.
These results do not rule out the other 44 analysed genes from playing a role in DCM, but they suggest that their contribution will be negligible or confined to very specific variant classes. This data will be instrumental to DCM genetic testing, both in the composition of diagnostic gene panels and in the interpretation of variants detected in patients, and to community gene curation efforts such as the one carried out by the ClinGen consortium, evaluating also other lines of evidence.
Post by Francesco Mazzarotto