The Cardiovascular Genetics and Genomics group is delighted to share the results of a new, multi-centre study on left-ventricular noncompaction (LVNC), published in Genetics in Medicine here.
LVNC is characterized by a partially noncompacted heart muscle, with a classically “spongy” appearance at imaging and diagnostic criteria based on the relative amount of compacted and noncompacted myocardium, as displayed in the figure below. Such diagnostic criteria though — when applied using the newer imaging technologies — can lead to the detection of LVNC in up to 15% of the population, highlighting a high risk of over-diagnosis.
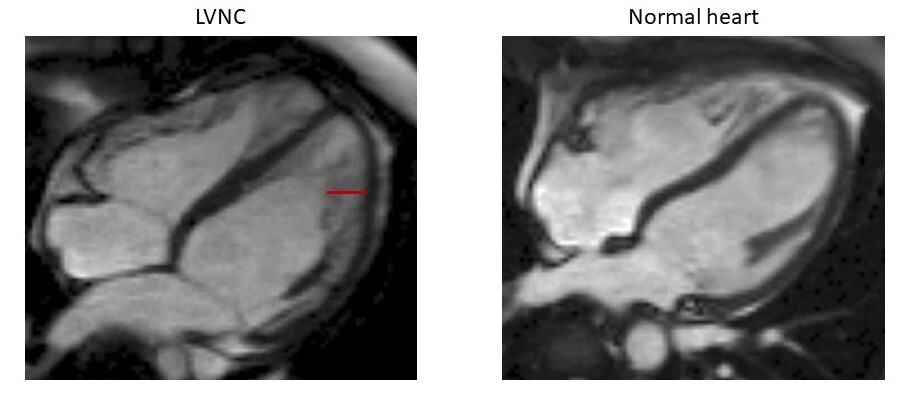
Hearts in 4-chamber view of a patient affected with LVNC (left) and a healthy individual (right), obtained with cardiac magnetic resonance imaging. The non-compacted portion of the myocardial layer in the patient with LVNC is highlighted with a red line.
The true nature of LVNC as a clinical entity is subject to much debate as to LVNC represents a separate disease or whether it is to be considered a secondary trait that can manifest in presence of an underlying cardiomyopathy. Such discussion is still far from reaching a definitive answer, as reflected by the two different classifications assigned by the European Society of Cardiology and the American Heart Association, which label LVNC as an “unclassified cardiomyopathy” and a “primary genetic cardiomyopathy”, respectively.
In this study, developed in collaboration with Amsterdam UMC, the University of Florence, the Aswan Heart Centre and the Partners’ Healthcare Laboratory for Molecular Medicine, we have:
- collected genetic data from over 800 patients referred for genetic testing for LVNC with LVNC (575 from previously published cohorts and 264 unpublished) and compared the frequencies of rare (carried by fewer than 1 in 10000 individuals) coding variants in 70 genes with those observed in a publicly available reference population (Genome Aggregation Database [gnomAD], ~125,000 individuals): this comparison showed which genetic variant classes are significantly associated with LVNC.
- compared results with those obtained utilizing the same strategy on cohorts of patients with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), in previous studies performed by our group: this analysis’ main purpose was to assess the genetic overlap between LVNC and these two other cardiomyopathies, so to estimate the proportion of cases in which non-compaction may be a secondary trait of underlying DCM or HCM, rather than a distinct process.
- Analyzed cardiac phenotypes of population individuals carrying specific LVNC-associated variants, so to assess the effect of such variants in the population and investigate whether their presence correlates with sub-clinical phenotypes: this genotype-phenotype analysis serves to understand if genetics can help discern between physiological and pathological noncompaction.
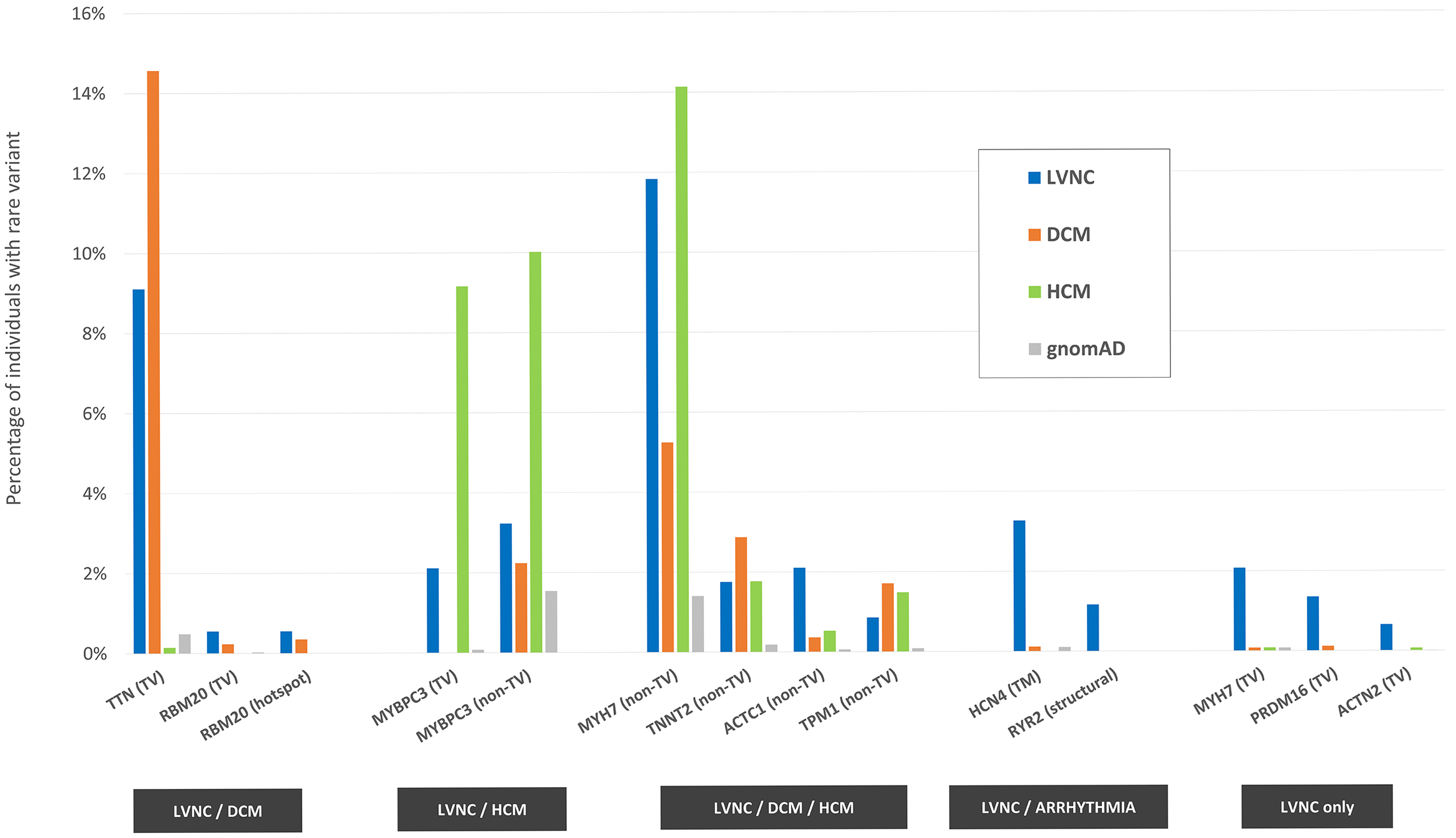
One figure from the paper, recapitulating the main results: 14 variant classes (in 11 genes) are associated with LVNC, having been found significantly more often in LVNC patients compared with population individuals (% indicated by the blue bars vs % indicated by the gray bars). Such variant classes are estimated to explain approximately 37% of cases. The disease association for the majority of them (11, in 9 genes) is shared with DCM (first group from the left, orange bars), HCM (second group, green bars) or both (third group). The associations with variants in the HCN4 and RYR2 genes are shared between LVNC and arrhythmogenic conditions, while the associations with truncating variants in MYH7, PRDM16 and ACTN2 are unique to LVNC. TV=truncating variants, TM=transmembrane, hotspot=residues 634-638 in RBM20.
As shown in the figure, the vast majority of LVNC-associated variant classes are also causative of HCM and/or DCM, suggesting how in a majority of cases LVNC is probably to be considered a secondary trait manifesting in patients affected with an underlying primary cardiomyopathy. However, there are certain cases (estimated in approximately 1 in 20 in the analyzed cohorts) in which LVNC is observed in presence of genetic variants that are uniquely LVNC-associated, suggesting how LVNC can also present as an isolated, distinct disease entity.
The association shared between LVNC and arrhythmogenic conditions with variants in the ion channel genes HCN4 and RYR2 (associated with sinus node disease and catecholaminergic polymorphic ventricular tachycardia, respectively) highlights how a subset of LVNC patients may be at risk of life-threatening arrhythmogenic events. Systematic screening for variants in these genes in LVNC patients may aid the early identification of such cases.
Of the variant classes associated uniquely with LVNC, truncating variants in MYH7 are of particular interest, as heterozygous “loss-of-function” variants have not previously been associated with cardiomyopathies (in contrast with non-truncating variants in the same gene, which are among the most important genetic causes of HCM and DCM). In this study, besides the significant association of these variants with LVNC, we also found how individuals in the general population (including the UK Biobank) carrying this type of variants have a significantly higher degree of ventricular noncompaction compared with non-carriers.
Taken together, these results contribute to the characterization of LVNC and suggest how:
- in the majority of cases – LVNC is a morphological phenotype manifesting in presence of an underlying cardiomyopathy.
- in approximately 5% of the cases, LVNC appears to be caused by a distinct genetic aetiology which suggests it can also be a distinct disease entity.
- in addition, results obtained on phenotyped population cohorts confirm a concrete danger of LVNC over-diagnosis, suggesting how diagnostic criteria for LVNC should not be based solely on imaging, but conjugated e.g. with genetic screening to help identify individuals at risk of developing left-ventricular dysfunction.
Written by Francesco Mazzarotto