We are pleased to share our Letter to the Editor published in Genetics in Medicine (McGurk et al. 2022) in response to the latest American College of Medical Genetics & Genomics (ACMG) recommendations on the reporting of secondary findings from clinical sequencing data (ACMG SF) (Miller et al., 2021 a, b).
The increasing availability of genome sequencing in clinical practice provides opportunities for improvements in health. One specific opportunity is through the early detection of health risks that might allow for preventative intervention. Since 2015 the ACMG has recommended that whenever clinical genome sequencing is undertaken, a defined set of genes should be actively interrogated for DNA variants that might herald a preventable clinical risk, such as a strong inherited predisposition to a specific cancer.
This is a recommendation for a genetic screening programme. Before implementing screening, it is necessary to understand the benefits and harms of the screening test, and the benefits and harms of any downstream interventions triggered by a positive test. It is also important to understand the costs of the programme, and who will bear these costs.
Genes associated with inherited cardiac conditions, including cardiomyopathies (heart muscle disease), make up about half of the latest ACMG SF gene list. These conditions can lead to sudden cardiac death, and sadly a fatal heart rhythm problem is sometimes the first manifestation of disease. Identifying people with these conditions early, before they even develop symptoms, is the only way of preventing such deaths. This is the motivation for screening when genetic data become available through testing for an unrelated condition.
However, not everybody who carries a “disease-causing” variant in a cardiomyopathy gene will develop disease. We call this incomplete penetrance. And at present there is huge uncertainty around this – when someone in the population is found to have a “disease-causing” variant, we don’t really know what risk they have of developing cardiomyopathy, and/or of having a dangerous heart rhythm.
We have no doubt that some people will benefit from screening, but others will also be harmed, e.g. through psychological harms, a loss of time or money due to burdensome surveillance, and potentially genetic discrimination. We do not believe that we have sufficient understanding of the relative benefits and harms to recommend that this should be carried out as standard-of-care.
Patients should be advised of this uncertainty. If a patient chooses to be informed of secondary findings this should ideally be done in the context of research to understand the benefits and harms.
We were particularly motivated to address this following recent update to the list of genes recommended for screening. The ACMG have added TTN (titin) and FLNC (filamin C) to the list of cardiomyopathy genes to evaluate. We have been studying Titin for some years, and we think that the inclusion of this gene presents huge challenges. Variants that we would call “likely pathogenic” if found in someone with cardiomyopathy are actually found in about 1 in 200 people in the general population. The vast majority never develop disease. Is there a net benefit in looking for these variants? We used data from the UK biobank to estimate what the benefits of screening might be.
Secondary findings in FLNC
The authors of the new recommendations say that FLNC variants are highly penetrant, conferring a high risk of cardiomyopathy and arrhythmia. We agree that people with cardiomyopathy due to variants in FLNC are more likely have rhythm problems than people with some other forms of cardiomyopathy. However, it has not been shown that these variants are highly penetrant when found in people in the general population.
In this analysis we studied participants in the UK Biobank (UKBB) and found 50 individuals with rare heterozygous FLNC-truncating variants (a similar prevalence to that seen in consortium populations) that would meet criteria for a P/LP annotation and might be returned as secondary findings. None of these individuals were apparently diagnosed with cardiomyopathy. The benefits of screening for these variants are unclear.
Secondary findings in TTN
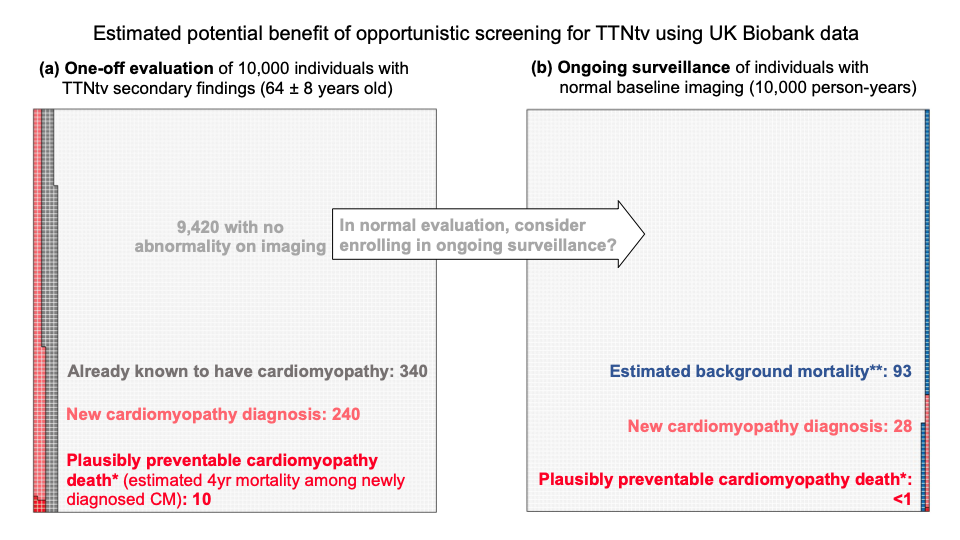
0.44% of UKBB participants carried rare (heterozygous) TTN-truncating variants, TTNtv. 1.4% of these were known to have cardiomyopathy when they signed up for the biobank study. These are therefore not strictly secondary findings.
A further 2.4% (1 in 40) showed evidence of a previously unrecognised cardiomyopathy on a one-off cardiac imaging assessment (derived from the proportion of heterozygous who met criteria for DCM during the UKBB cardiac MRI – CMR). This gives us an estimate of the number of people we would diagnose with cardiomyopathy if we offered people with SFs in TTN a one-off assessment.
Further individuals developed cardiomyopathy over time. To detect these would require ongoing surveillance, likely with CMR repeated at intervals. We estimate that this would detect a further ~3 incident cases per 1,000 person-years of ongoing surveillance in the population of people found to have TTN variants as secondary findings (200 people under surveillance for 5 years each = 1000 person-years).
Modelling adverse outcomes
So far, we have looked at how many people will develop cardiomyopathy. What we are really interested in is how many people will run into problems that might be preventable by early detection and intervention, particular sudden death.
Based on previous estimates that about 1 in 25 people with established dilated cardiomyopathy might die suddenly over a four-year window, we estimated that to prevent one death we would need to make 25 new diagnoses of cardiomyopathy, which would require us to carry out ~8,000 person-years of surveillance in people found to have TTN variants as secondary findings.
In other words, if we called back 1600 people who had a normal CMR, and repeated the test 5 years later, we might find 25 people who had now developed DCM, and treating these might prevent a single sudden death.
This assumes that the death is entirely preventable with early diagnosis and treatment, which is likely over-optimistic. Moreover, it ignores the fact that some of these people will not benefit from screening because they have other more immediate health concerns – e.g., they may have had genetic testing because of a cancer, or another rare genetic condition.
An alternative methodology (based on the total observed excess mortality amongst TTN variant carriers, and optimistically assuming that this excess mortality is entirely preventable) suggests that we would need to enrol 100 people into long-term surveillance to prevent 1 death in 10 years.
We therefore believe that while some people will benefit from return of secondary findings, it is premature to recommend this as standard-of-care until we have a better understanding of the benefits and harms of such screening.
Response to McGurk et al.
The authors of the ACMG recommendations responded to our letter (Gollob et al. 2022). They pointed out that the UKBB cohort may underestimate disease prevalence and incidence. We acknowledge the likelihood of survivorship bias in the UKB in our letter but note that the prevalence of DCM is very close to other population estimates, so do not believe that this bias has distorted our estimates importantly.
Furthermore, the UKB cohort likely provides very reasonable estimates for opportunistic screening carried out in adults, e.g., for adult cancer patients, or healthy adults undergoing clinical sequencing because they have a child with a rare genetic condition (for trio analyses).
The authors agreed that more studies are needed on the costs and benefits of reporting SFs and add that they can adapt the future gene lists as new evidence becomes available.
References
- Gollob MH, Hershberger RE, Gordon AS, et al. Response to McGurk et al. Genet Med. 2022;24(3):747-748.
- McGurk KA, Zheng SL, Henry A, et al. Correspondence on “ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG)” by Miller et al. Genet Med. 2022;24(3):744-746.
- Miller DT, Lee K, Gordon AS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1391–1398.
- Miller DT, Lee K, Chung WK, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1381–1390. Published correction appears in Genet Med. 2021;23(8):1582-1584.