Determining the clinical impacts of genetic variation is crucial to fully realizing the promise of genome medicine. Missense variants, which alter just one amino acid in protein sequences, are particularly challenging to interpret. With the approximately 70 million possible missense variants in the human genome, the clinical impact of the majority (>94%) remains unknown. Our study Zhang et.al 2024, published in Genome Medicine, introduces a novel computational approach called Homologous Missense Constraint (HMC). HMC measures genetic intolerance at the amino acid level within protein domains, significantly enhancing the prioritization of pathogenic missense variants and facilitating disease gene discovery. Here, we summarize the highlights of our study.
Novel methodology to prioritise missense variants
Existing computational methodologies to prioritize missense variants include predicting variants under negative selection based on cross-species conservation, structural impact prediction, protein language modelling, genetic constraints within human populations and combining multiple of the above strategies. Genetic constraint measures, which assess genetic intolerance in human populations, are emerging as a promising approach. If a genetic variant causes severe disorders, such as affecting reproductive fitness, we expect it to be depleted within human populations. With the growing catalogue of naturally occurring human variations, we can assess the degree of this depletion, making genetic constraints powerful tools for gene and variant prioritization. They have also been instrumental in helping us interpret non-coding regions.
However, existing missense constraint methods are limited by resolution, assessing either entire genes or large sub-genic regions. Given the current size of the human exome aggregation database (gnomAD v2 125K exomes), we are still underpowered to evaluate the depletion of variants at individual residues. Instead of measuring constraint at individual amino acids, our approach HMC evaluates homologous residues in protein domains to aggregate the genetic constraint signal.
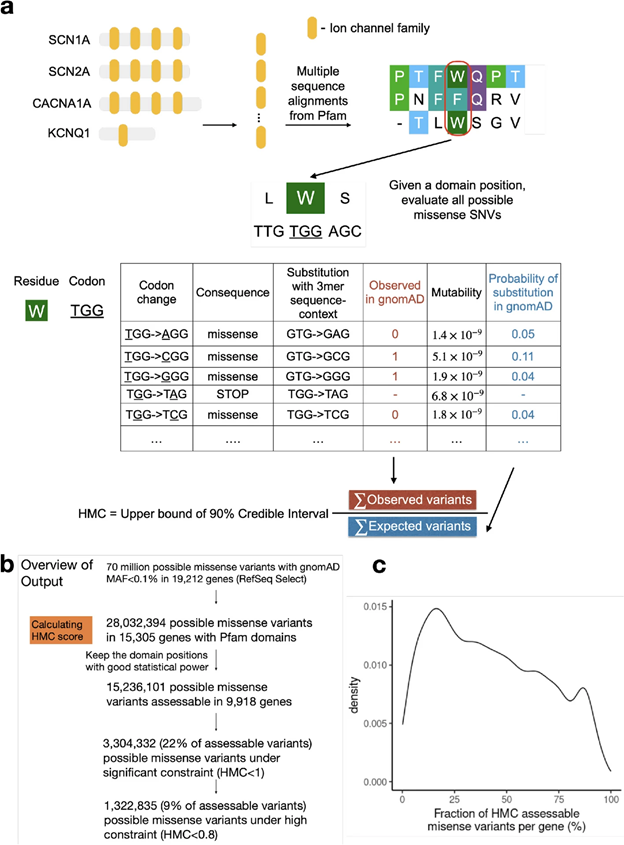
Fig1. Illustration of the HMC method
Robust benchmarking shows that HMC precisely predicts pathogenic missense variants and is complementary to existing pathogenicity tools
Although HMC can only be applied to 22% of the exome, we show that it is a highly precise metric for predicting deleterious missense variants for both early-onset and adult-onset disorders. Its precision is comparable to state-of-the-art supervised meta-predictors, though with low sensitivity. We applied HMC to prioritize deleterious de novo mutations in individuals with developmental disorders, demonstrating that HMC outperforms existing tools and identified seven newly-significant developmental disorder genes. Finally, we demonstrate that HMC is orthogonal to many existing variant prioritization measures and complementary to existing gene-level or sub-genic measures of genetic constraint.
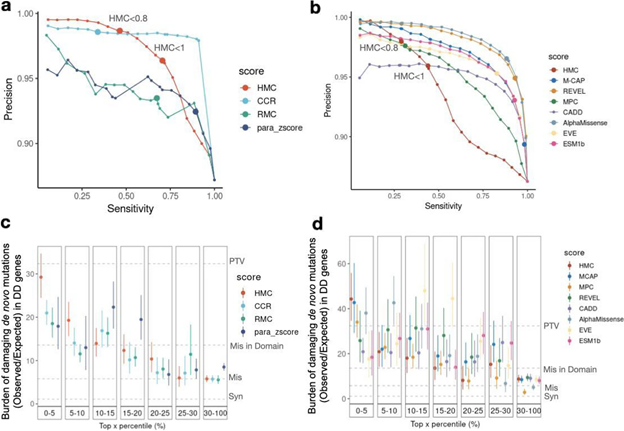
Fig2. Benchmarking HMC with existing pathogenicity scores
Recommended usage
We have made HMC scores available online via cardiodb.org and the “Constraint scores” track in the UCSC genome browser. Given its high precision, we recommend using HMC in clinical interpretation under the framework of ACMG guidelines. HMC can be used as a constraint metric applied through PP2 (PP2: “Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease”). We suggest evaluating PP2 by using HMC first where possible (activating PP2 if HMC < 0.8) before applying gene/region-level constraint as illustrated below (Fig 3). To combine HMC with machine learning pathogenicity predictors or other lines of evidence, we recommend following the ACMG guidelines for combining criteria to classify variants. Since lack of constraint indicates a lack of evidence of pathogenicity, we do not recommend using HMC unconstrained prediction as evidence of benign impact.
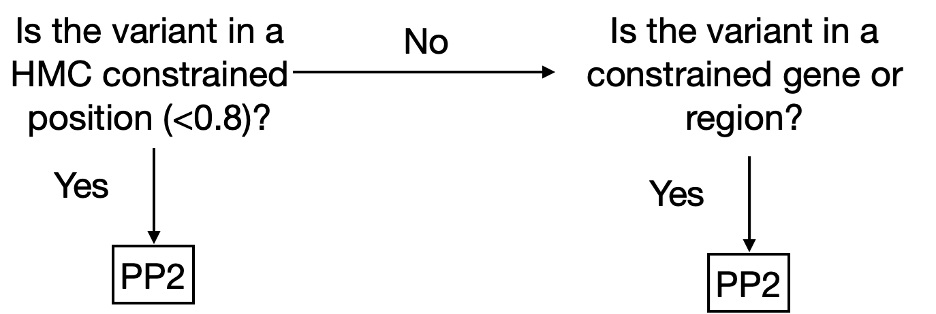
Fig. 3. Decision tree to use HMC score in a clinical workflow as PP2 (supporting evidence of pathogenicity) following the ACMG guidelines.
By focusing on genetic constraints at the single amino acid level within protein domains, HMC provides a highly precise tool for predicting pathogenic variants and discovering new disease genes.