The latest ClinGen curation of hypertrophic cardiomyopathy (HCM) genes is now available in pre-print! You can read the full details here.
There are four new definitive HCM genes: ACTN2, CSRP3, FHOD3, and TNNC1.
We have added these to our CardiacG2P dataset. You can check out the latest version of CardiacG2P directly on our github repo, on our dedicated documentation page and on the official EBI G2P page.
Just like in previous releases, this update includes key information on inheritance patterns, allelic requirements, disease-associated variant consequences, and variant classes for each gene-disease pair.
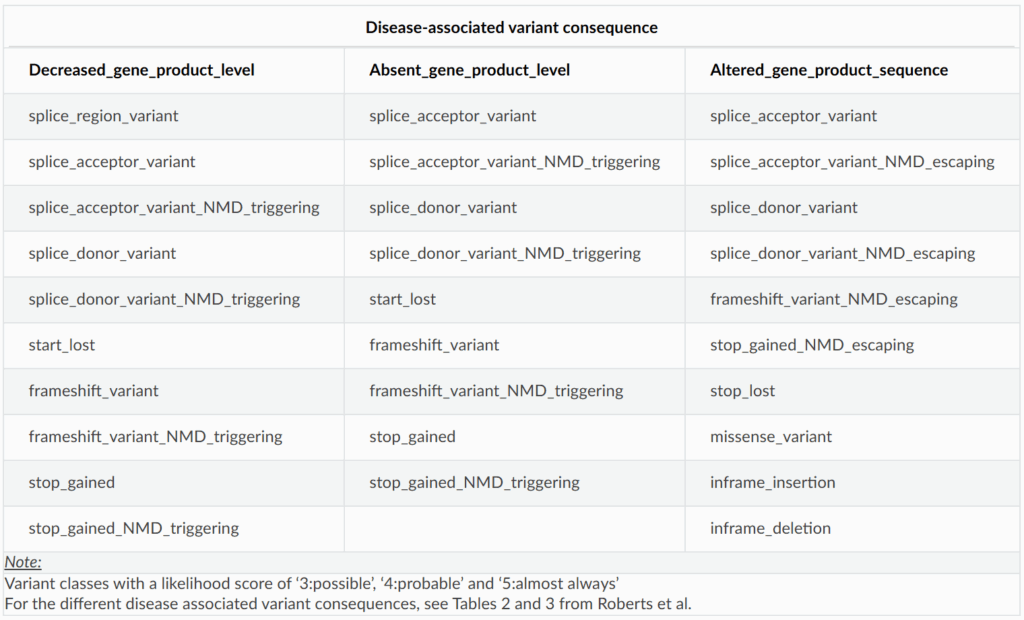
Which Variant Classes Should You Filter For?
The structured dataset lists variant classes that have been reported to cause disease for each gene-disease pair. Importantly, we expect that other variant classes with similar sequence level consequences could also cause disease. For example, if frameshift variants that trigger nonsense-mediated decay (NMD) are known to cause disease, then NMD-triggering stop gained variants may be disease causing as well.
To streamline variant prioritisation, we recommend filtering for all variant classes that map to the disease-associated variant consequences for a given gene-disease pair. Here’s a useful breakdown of variant consequences and their corresponding variant classes:
Use this table to guide your variant filtering strategy
For instance, if variants in a gene cause disease through decreased gene product level and stop gained variants have been implicated, you should also filter for variants like splice donor and frameshift variants.
For a more detailed explanation of the disease-associated variant consequences refer to Tables 2 and 3 in Roberts AM et al 2024 (PMID: 37982373). These tables outline the predicted impact of different variant classes at the sequence level.
We are excited to share our publication (Jennings et al. 2025) in Cardiology in the Young describing discussion needs and potential research avenues for young people with dilated cardiomyopathy (DCM). We hope it is a valuable insight for clinicians and researchers, particularly for the ongoing development of clinical care specifically for the young with DCM.
DCM is a heart condition characterised by an enlarged left ventricle that can lead to heart failure. It is most often diagnosed in adult males, however, when diagnosed in childhood, it can present unique challenges to patients and clinicians. As children move from the paediatric hospital services to the adult services (“transition”), they need unique support when taking responsibility for their health and living with DCM.
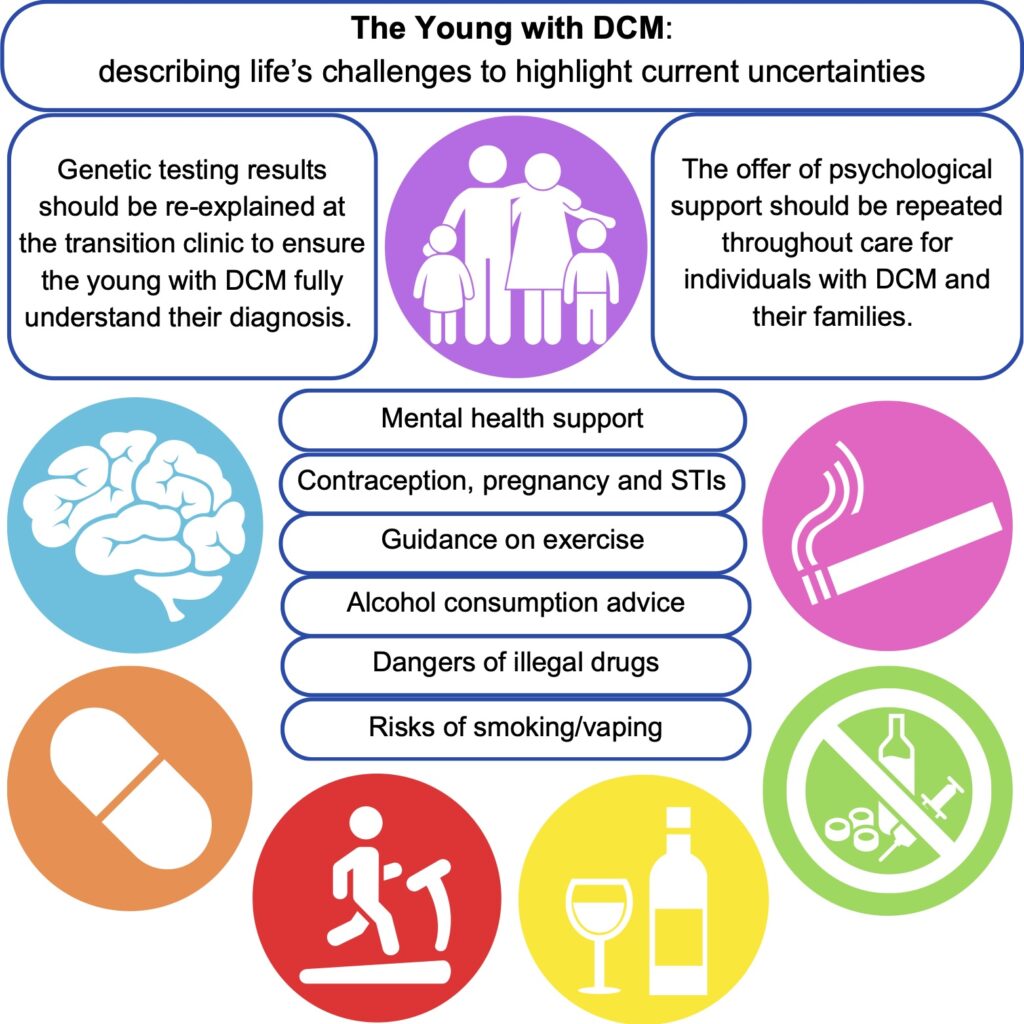
Using a patient-scientist’s perspective to inform a literature review, we identified areas that could form the basis of the information provided during transition clinical services. This could aid the further standardisation of clinical care for young people and ensure that the correct support is in place to equip young people to make informed decisions and provide an opportunity for discussion about topics that may be experienced when living with a chronic condition. The key areas identified as requiring additional support from clinical teams are: mental health, exercise, alcohol consumption, cigarettes, recreational drugs, and sexual health.
Young people with DCM and their families require specialised mental health support to equip them for lifelong clinical care. For patients, it is important to support them as they come to terms with their limitations amongst healthy peers. Family members may have experienced traumatic events such as sudden death or feel the empathetic burden of being the “well” sibling. Medical trauma during childhood can impact stress tolerance in later life and young people may be more susceptible to poor mental health outcomes without intervention. Repeated signposting to mental health services is recommended for both the patient and their families.
Genetic testing is often undertaken for diagnostic support in cardiomyopathies. If carried out during childhood, results should be revisited with the young person in the transition to adulthood. This would ensure the patient has a full understanding of the downstream effects of a given result and the impact on access to family planning measures, etc.
Current exercise guidelines suggest it is safe to exercise to perspiration with the ability to hold a conversation and avoid exercising when feeling unwell or tired. The advice however is generally non-specific regarding varying exercise types and duration, with little research available on DCM in the young.
Alcohol and caffeine are regularly consumed in adults, but the evidence surrounding their negative impacts is non-specific, and the advice is to avoid them in excess, hydrate more before and after consumption, and avoid taking them in combination. They may oppose the effects of therapeutic drugs used for DCM treatment.
Smoking, vaping, cannabis, cocaine, MDMA, and ketamine, negatively influence the cardiovascular system, with cocaine described as the most cardiotoxic. There is little specific evidence for patients with cardiomyopathy, and it is recommended by medical professionals to avoid consumption. When advocating abstinence to teenagers, it is important to discuss the reasoning for this.
Pregnancy can be contraindicated in some people with DCM, and progesterone-only or barrier methods of contraception are recommended to avoid the risks associated with oestrogen-based alternatives. During the transition to adult services, the implications and planning of pregnancy should be discussed to ensure that patients are well-informed about the importance of avoiding unplanned pregnancies.
Further research is needed to address the many uncertainties in these areas with respect to young age, particularly for physical activity, and such guidance would be welcomed by the young with DCM who must come to terms with being different and more limited amongst healthy peers.
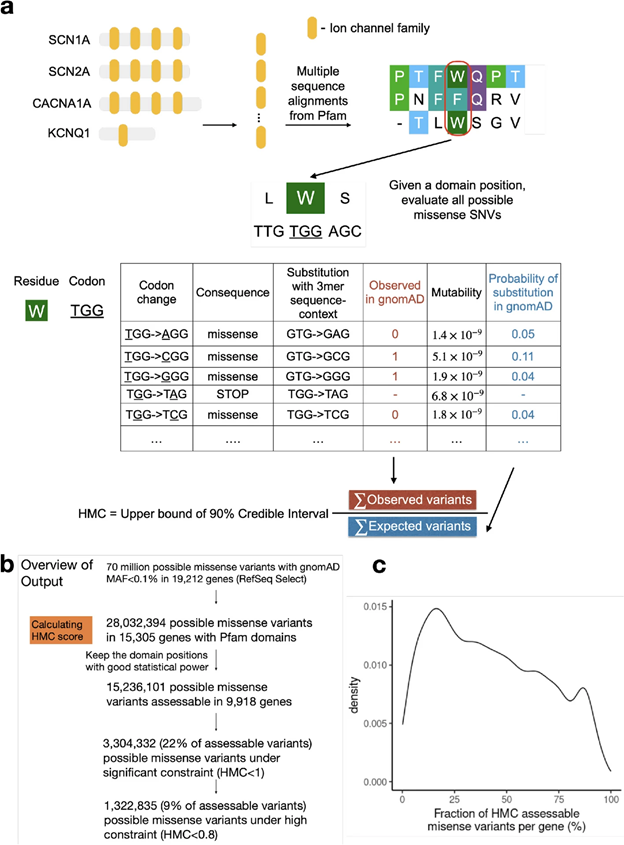
Determining the clinical impacts of genetic variation is crucial to fully realizing the promise of genome medicine. Missense variants, which alter just one amino acid in protein sequences, are particularly challenging to interpret. With the approximately 70 million possible missense variants in the human genome, the clinical impact of the majority (>94%) remains unknown. Our study Zhang et.al 2024, published in Genome Medicine, introduces a novel computational approach called Homologous Missense Constraint (HMC). HMC measures genetic intolerance at the amino acid level within protein domains, significantly enhancing the prioritization of pathogenic missense variants and facilitating disease gene discovery. Here, we summarize the highlights of our study.
Novel methodology to prioritise missense variants
Existing computational methodologies to prioritize missense variants include predicting variants under negative selection based on cross-species conservation, structural impact prediction, protein language modelling, genetic constraints within human populations and combining multiple of the above strategies. Genetic constraint measures, which assess genetic intolerance in human populations, are emerging as a promising approach. If a genetic variant causes severe disorders, such as affecting reproductive fitness, we expect it to be depleted within human populations. With the growing catalogue of naturally occurring human variations, we can assess the degree of this depletion, making genetic constraints powerful tools for gene and variant prioritization. They have also been instrumental in helping us interpret non-coding regions.
However, existing missense constraint methods are limited by resolution, assessing either entire genes or large sub-genic regions. Given the current size of the human exome aggregation database (gnomAD v2 125K exomes), we are still underpowered to evaluate the depletion of variants at individual residues. Instead of measuring constraint at individual amino acids, our approach HMC evaluates homologous residues in protein domains to aggregate the genetic constraint signal.
Fig1. Illustration of the HMC method
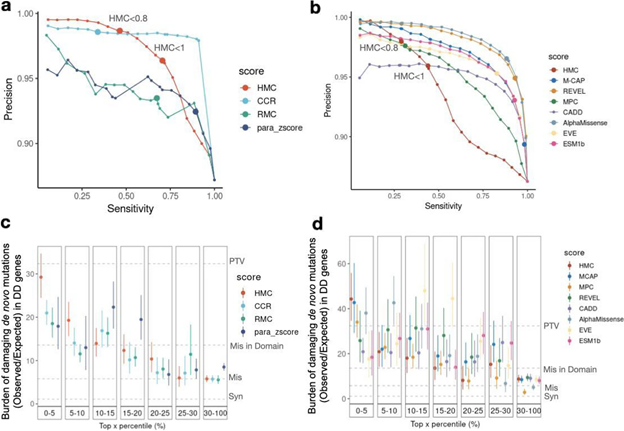
Robust benchmarking shows that HMC precisely predicts pathogenic missense variants and is complementary to existing pathogenicity tools
Although HMC can only be applied to 22% of the exome, we show that it is a highly precise metric for predicting deleterious missense variants for both early-onset and adult-onset disorders. Its precision is comparable to state-of-the-art supervised meta-predictors, though with low sensitivity. We applied HMC to prioritize deleterious de novo mutations in individuals with developmental disorders, demonstrating that HMC outperforms existing tools and identified seven newly-significant developmental disorder genes. Finally, we demonstrate that HMC is orthogonal to many existing variant prioritization measures and complementary to existing gene-level or sub-genic measures of genetic constraint.
Fig2. Benchmarking HMC with existing pathogenicity scores
Recommended usage
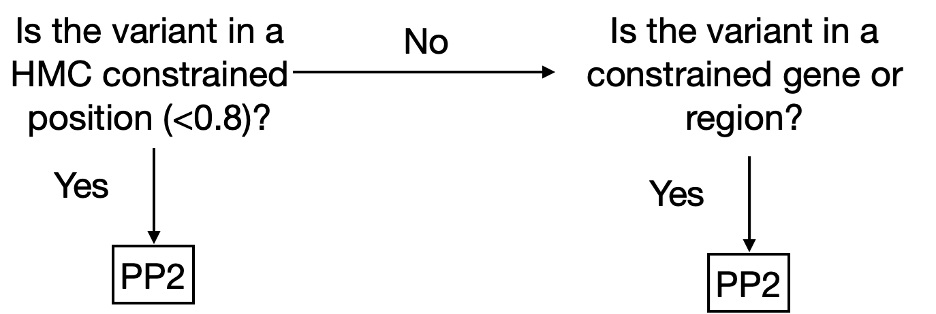
We have made HMC scores available online via cardiodb.org and the “Constraint scores” track in the UCSC genome browser. Given its high precision, we recommend using HMC in clinical interpretation under the framework of ACMG guidelines. HMC can be used as a constraint metric applied through PP2 (PP2: “Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease”). We suggest evaluating PP2 by using HMC first where possible (activating PP2 if HMC < 0.8) before applying gene/region-level constraint as illustrated below (Fig 3). To combine HMC with machine learning pathogenicity predictors or other lines of evidence, we recommend following the ACMG guidelines for combining criteria to classify variants. Since lack of constraint indicates a lack of evidence of pathogenicity, we do not recommend using HMC unconstrained prediction as evidence of benign impact.
Fig. 3. Decision tree to use HMC score in a clinical workflow as PP2 (supporting evidence of pathogenicity) following the ACMG guidelines.
By focusing on genetic constraints at the single amino acid level within protein domains, HMC provides a highly precise tool for predicting pathogenic variants and discovering new disease genes.
We are pleased to share our publication (McGurk et al. 2024) in Circulation on the pharmacogenetic influences over mavacamten pharmacokinetics.
Mavacamten is a first-in-class, orally administered, cardiac-specific, small-molecule allosteric modulator of β-cardiac myosin. Mavacamten reversibly inhibits the binding of β-cardiac myosin to actin to reduce hypercontractility in an exposure-dependent manner.
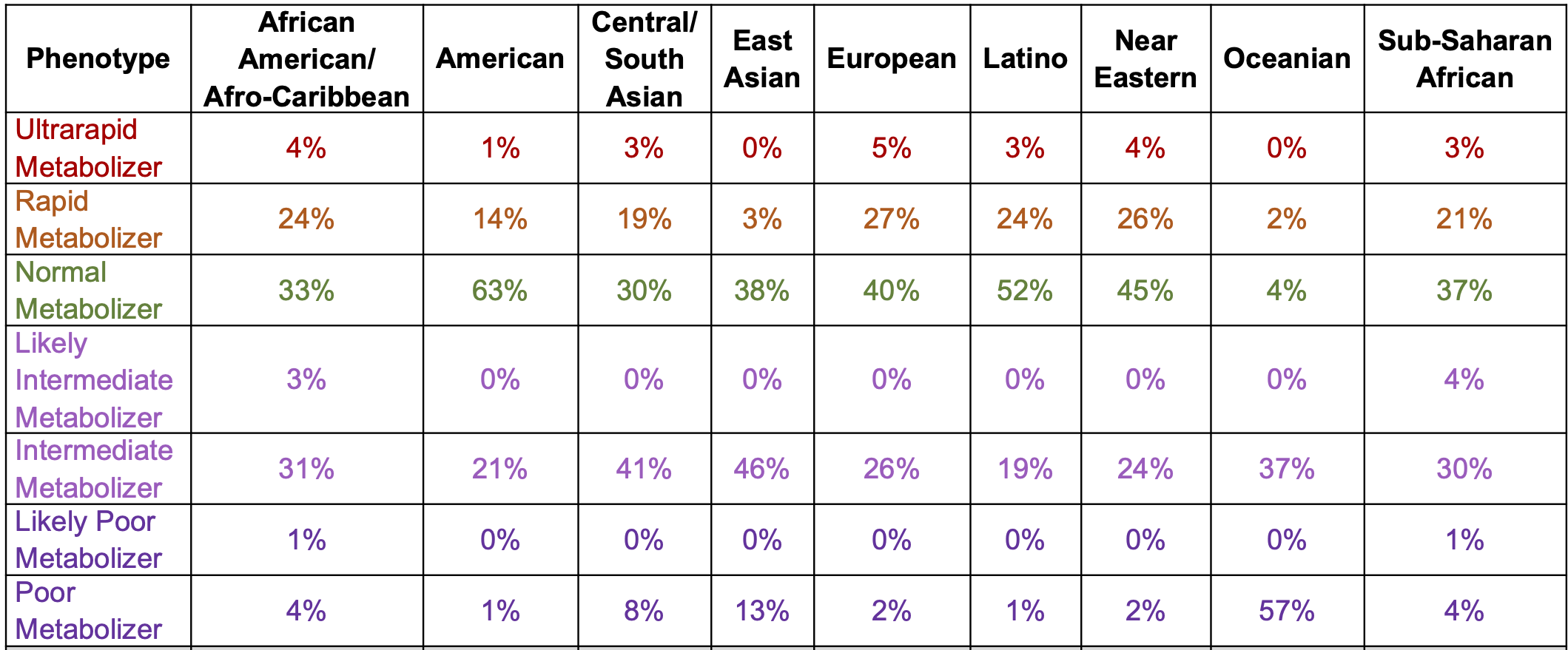
DNA variants can alter the therapeutic and adverse effects of drugs at recommended dosages. Variants in genes encoding cytochrome P450 enzymes, expressed in the liver and small intestine, are associated with variable drug metabolism. Individuals can be categorized into 5 metabolizer phenotypes: poor, intermediate, extensive (sometimes described as normal), rapid, and ultrarapid. CYP2C19 is the dominant metabolizer of mavacamten and poor metabolizers, attributable to diplotypes of 2 CYP2C19 alleles [the CYP2C19*2 (p.Pro227Pro;c.681G>A;rs4244285) and CYP2C19*3 (p.Trp212Ter;c.636G>A;rs4986893) alleles], are at increased risk of systolic dysfunction from mavacamten treatment at the recommended dose. Individual metabolizer status is further complicated by additionalvariation across other common alleles within this gene, other CYP450 genes, and other relevant pathways.
The prevalence of poor metabolizers (PMs) is common and varies by population (below). The half-life is extended to 23 days in PMs from 9 days in normal metabolisers (NMs). In NMs, it takes ~4 weeks (five half-lives) to eliminate mavacamten from the body after treatment discontinuation.
The EMA and UK Medicines and Healthcare Products Regulatory Agency recommend genotyping for CYP2C19 to determine the appropriate dose. If treatment is initiated without metabolizer status determination, dosage should follow as described for poor metabolizers (starting at 2.5 mg once daily and a maximum dose of 5 mg once daily). The recommended starting dose for all other metabolizers is 5 mg once daily and a maximum dose of 15 mg once daily. Dose modifications are provided for concomitant medicinal products, including CYP2C19 and CYP3A4 inhibitors and inducers. The EMA and Australian Therapeutic Goods Administration suggest a simulated 5 mg dose in a poor metabolizer is similar to the maximum dose (15 mg) in a normal metabolizer.
It remains unclear how the EMA genotyping recommendation will be implemented across diverse European health care systems. Genetic testing for variants in sarcomere-encoding genes is used for individuals with HCM to establish the molecular etiology; pharmacogenetic analysis could be incorporated for individuals who have not already undergone testing and allow for the EMA recommended dosage stratification.
The NICE guidelines note uncertainty surrounding the impact of sarcomeric variants on treatment effect (section 3.7): the relationship between treatment response and genotype has not been fully characterised, with the possibility of differences between individuals with and without sarcomere variants and/orwith variants in thick vs thin filament genes.
Providing the appropriate mavacamten dosage to each individual with oHCM from treatment initiation should allow for improved quality of life at the lowest risk of adverse events, cost, and burden to health care systems. Prescribers must be aware of the potential for metabolic variability across and within different ancestries and clinical vigilance with close monitoring will be required to avoid adverse events. Successful treatment requires improving symptoms of oHCM in rapid metabolizers and minimizing the risk of drug-induced systolic dysfunction in poor metabolizers. Treatment without genotyping risks reduced ejection fraction in poor metabolizers or increased time to therapeutic dose in normal metabolizers. With limited medications for the management of oHCM, or where there is limited access to septal reduction therapy, effective titration of cardiac myosin inhibitors is vital to the success of treatment. Whereas future clinical trials with improved metabolizer and ancestral representation will aid our understanding in this area, CYP2C19 genotyping may allow for less frequent clinical monitoring and reduced costs.
We also discuss i) only NICE recommends mavacamten as an add-on to standard care and notes long waiting times for echo; ii) Tian et al 2023 which studied 7 PMs; iii) the potential for predose mavacamten plasma concentration measurement (based on 21 individuals of phase 1 trial).
The NHS England National Genomics Education Programme has also released some guidance and an example clinical scenario
This is just one example of how common DNA variants influence cardiovascular treatment. Pharmacogenetic influences are known and reported for drugs during trials, but to date are used clinically only in a few areas. Further implementation in a cardiovascular setting would allow for reduced adverse events, time to therapeutic dose, and titration, of drug interventions.
The UK Rare Disease Research Platform has been established with a £14 million investment over five years by the Medical Research Council (MRC) and the National Institute for Health and Care Research (NIHR). It is made up of a central coordination and administrative hub and eleven specialist nodes based at universities across the UK.
The aim of the UK Rare Disease Research platform is to bring together expertise from across the UK rare disease research system to foster new and innovative treatments for those directly and indirectly impacted by rare conditions.
We are delighted to share our recent article (Allouba et al.) on the influence of ethnicity and consanguinity on the genetic architecture of hypertrophic cardiomyopathy.
Accurate interpretation of genetic variants identified in HCM patients represents a major challenge for diagnosis and implementing precision medicine, especially in understudied populations. Therefore, our aim was to define the genetic architecture of HCM in the largest Middle East and North African (MENA) cohort analysed to date by leveraging an ancestry-matched Egyptian case-control cohort recruited to the Aswan Heart Centre. We also compared genetic data between Egyptian and predominantly European patients to identify patterns of genetic variation that are unique to consanguineous populations of MENA ancestry and are likely to be more important contributors to HCM.
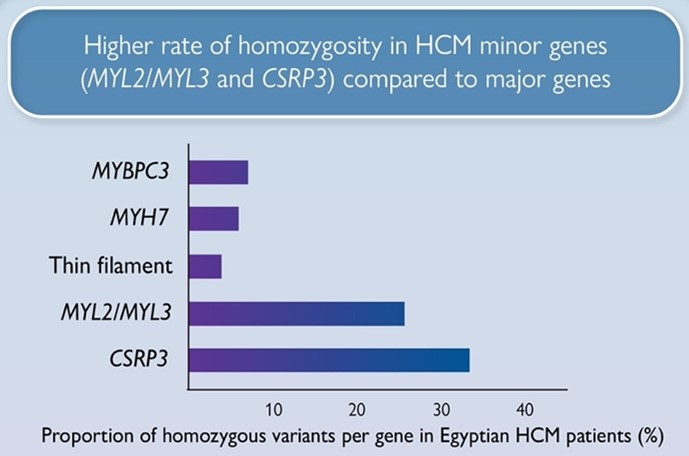
We report a markedly higher rate of homozygosity in HCM minor genes MLC- (MYL2, MYL3) and CSRP3 genes compared to major HCM genes (MYBPC3, MYH7), suggesting these variants have low penetrance in heterozygosity, but contribute to recessive disease. Along with the recently reported recessive HCM gene, TRIM63, these genes could be more relevant to consanguineous populations.
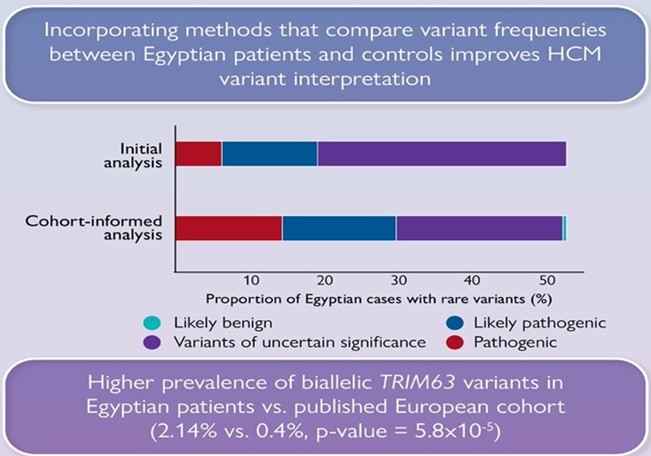
We also show that significantly fewer rare variants detected in Egyptian HCM patients could be classified as (likely) pathogenic compared to Europeans due to the underrepresentation of MENA populations in current HCM databases. Integrating methods that leverage Egyptian controls increased the yield of clinically actionable variants (pathogenic, likely pathogenic) from 19% (initial analysis) to 29.6% (cohort-informed analysis)
Collectively, these findings will enhance the utility of clinical genetic testing for understudied populations as well as our understanding of the genetic aetiology of HCM. Analysis of such highly consanguineous cohorts opens new research avenues for the discovery of novel recessive genes in future exome or genome sequencing studies
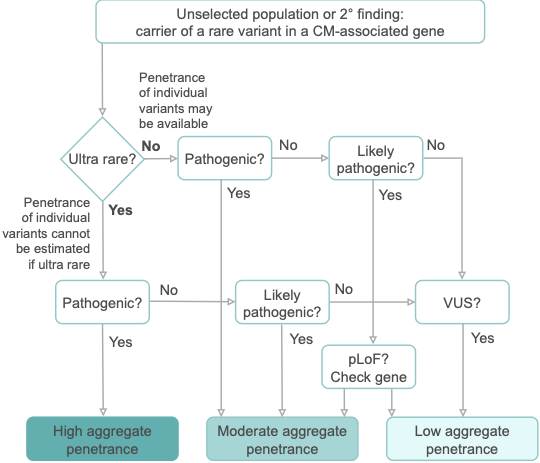
We are pleased to share our publication (McGurk et al. 2023) in the American Journal of Human Genetics on the penetrance of rare variants in cardiomyopathy-associated genes: a cross-sectional approach to estimate penetrance for secondary findings.
The penetrance of cardiomyopathies (CMs) is incomplete and age-related, and expressivity is highly variable. These features present huge challenges for disease management. In particular, the penetrance of individual variants in CM-associated genes is incompletely characterised and poorly understood, especially when identified in asymptomatic individuals without family history.
With the growing availability of whole exome sequencing in wider clinical settings and consumer-initiated elective genomic testing, the importance of estimating the penetrance of individual variants identified as secondary findings (SFs) to guide intervention is ever-increasing. Genes associated with inherited CMs make up one-fifth of the 78 genes recommended by the American College of Medical Genetics and Genomics (ACMG SF v3.1) for reporting SFs during clinical sequencing. Variant-specific estimates of penetrance are required to appropriately inform clinical practice and to fully utilise genetics as a tool to individualise the risk of developing disease in asymptomatic carriers.
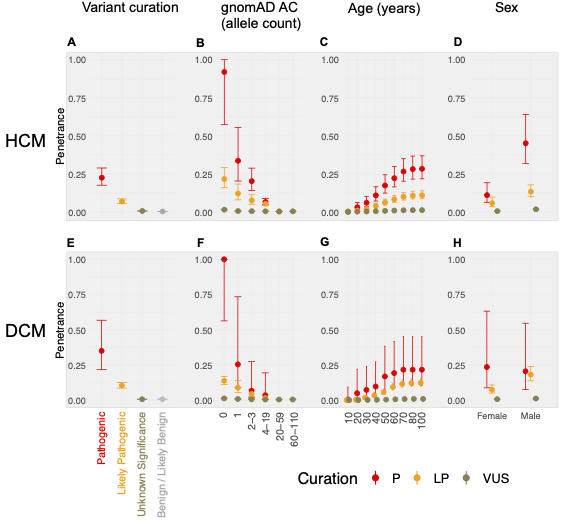
We apply a cross-sectional approach, using a method that compares the allele frequency of individual rare variants in large cohorts of cases and reference populations to estimate penetrance. Sequencing data for 10,400 individuals referred for HCM genetic panel sequencing and 2,564 individuals referred for DCM genetic panel sequencing were included in the analysis. To estimate the prevalence of CMs, a literature review and meta-analysis were undertaken, resulting in prevalence estimates for HCM (1:543; 1:1,300 women, 1:360 men) and DCM (1:220; 1:340 women, 1:160 men).
In aggregate, the penetrance by late adulthood of rare, pathogenic variants (23% for HCM, 35% for DCM) and likely pathogenic variants (7% for HCM, 10% for DCM) was substantial for dominant CM. Penetrance was significantly higher for variant subgroups annotated as loss of function or ultra-rare and for males compared to females for variants in HCM-associated genes.
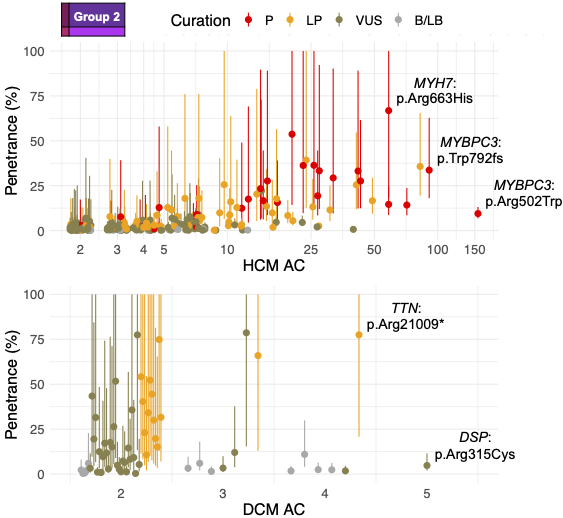
We estimated variant-specific penetrance for 316 recurrent variants most likely to be identified as SFs (51% HCM and 17% DCM cases). 49 variants were observed at least ten times (14% of cases) in HCM-associated genes. Median penetrance was 14.6% (±14.4% SD). We explore estimates of penetrance by age, sex, and ancestry, and simulate the impact of including future cohorts.
This dataset is the first to report the penetrance of individual variants at scale and will inform the management of individuals undergoing genetic screening for SFs. While most variants had low penetrance and the costs and harms of screening are unclear, some carriers of highly penetrant variants may benefit from SFs.
We are pleased to share a new article by Valentina Santofimio on research she completed during her masters programme with us.
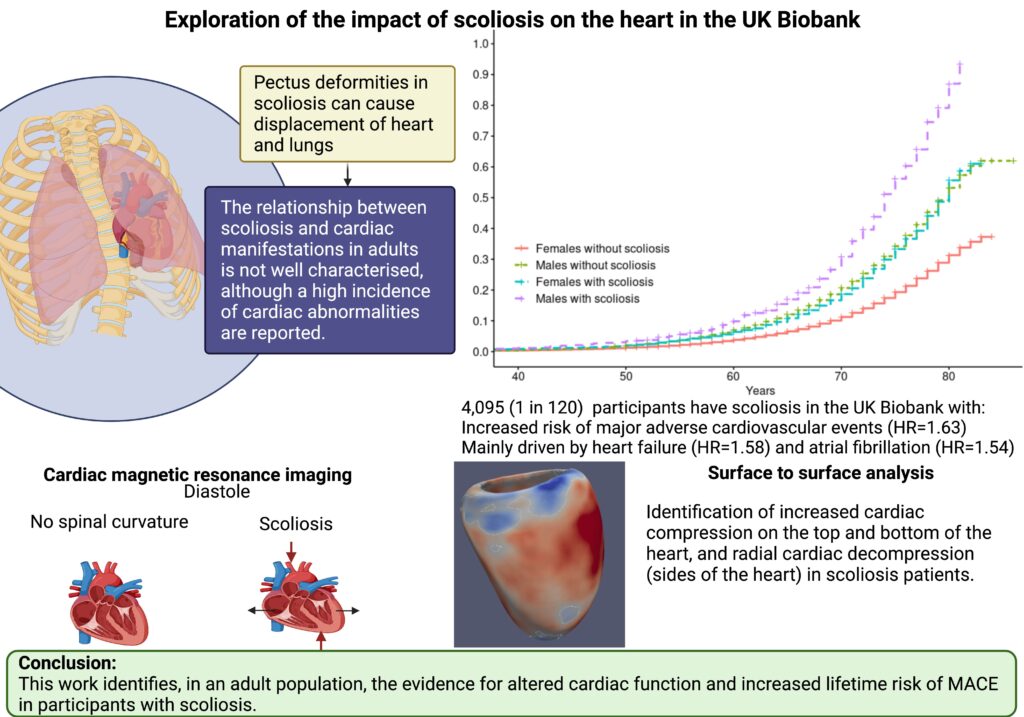
The abnormal curvature of the spine in scoliosis patients can impact organs within the ribcage including the heart. Most cardiac studies of scoliosis patients to date surround investigations into congenital heart disease. The relationship between scoliosis and non-congenital cardiac manifestations in adults is not well characterised.
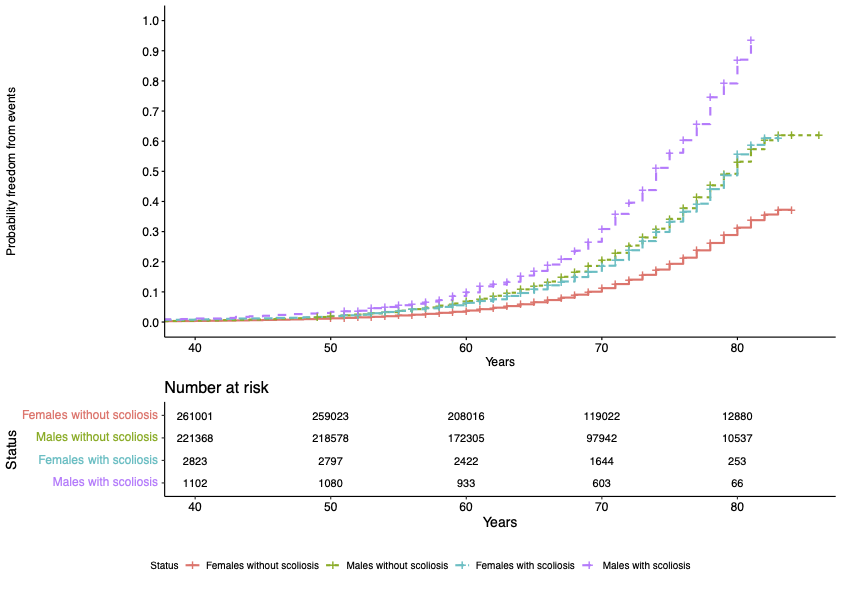
Our study focused on investigating the impact of scoliosis on the heart through assessment of cardiac MRI (CMR) traits in the UK Biobank (UKB) adult population cohort. A total of 4,095 (0.8%, 1 in 120) UKB participants were identified to have all-cause scoliosis.
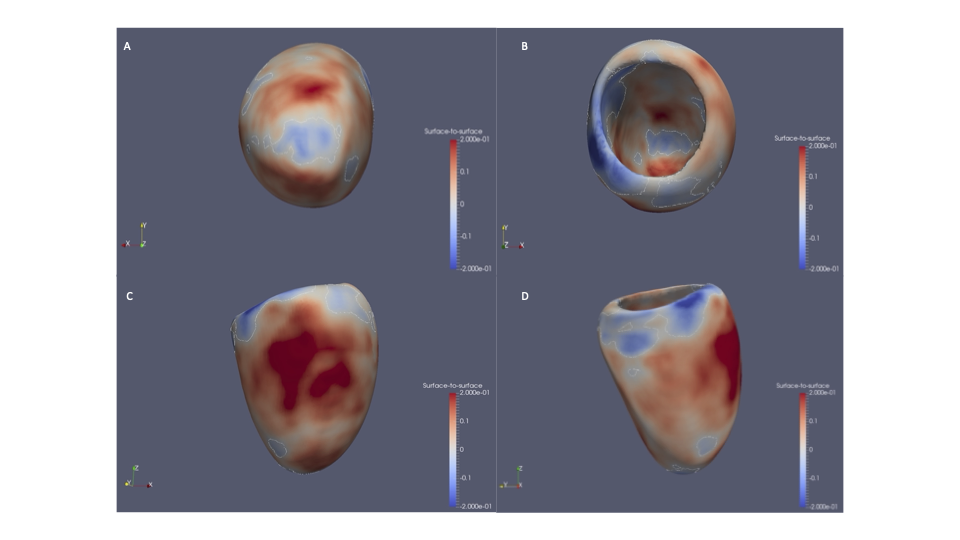
Significant associations were found between scoliosis and older age, female sex, heart failure, valve disease, hypercholesterolemia, diagnosis of hypertension, and decreased enrolment for CMR. We identified altered radial and longitudinal peak diastolic strain rates (PDSR) in participants with scoliosis with CMR available compared to participants without diagnosis of scoliosis. 3D cardiac modelling also showed altered cardiac strain.
A significantly increased lifetime risk of MACE was observed for UKB participants with scoliosis (HR=1.45, P<0.001), mainly driven by heart failure (HR=1.58, P<0.001) and atrial fibrillation (HR=1.54, P<0.001). The probability of MACE doubled in males into older age (from 60 years of age). This may be caused through the altered cardiac diastolic strain rates observed in participants with scoliosis.
The abnormal curvature of the spine can increase mechanical constraint on the heart which may result in diastolic dysfunction and the severity of the spinal deformity has been shown to aggravate ventricular and right atrial pressure.
Scoliosis may be an important modifier of cardiac strain in the adult population. This has clinical implications for the consideration of undertaking scoliosis treatment surgery. However, further research is required to follow up the role of scoliosis in cardiac manifestations in a clinical setting, alongside genetic analyses to assess causality.
Rare and extremely rare variants can be difficult to interpret, and at times genotype-phenotype associations can be hard to determine. DECIPHER is an interactive web-based database of genomic variants and associated phenotypes. It contains data from an international community of more than 270 centers and academic departments that focus on clinical genetics and rare disease genomics.
The web interface allows for depositing and sharing of genomic variants and phenotypes associated with an individual patient. Centers that deposit a variant can choose to keep data private, share with other members within a pre-defined consortium, or with patient consent can contribute to the open-access data that is available on DECIPHER.
DECIPHER project sharing has supported collaborations facilitating patient diagnoses, and the use of DECIPHER has contributed to over 2600 published articles since 2004.
DECIPHER was established in 2004 to enable the interpretation and sharing of genotype and phenotype data from rare disease patients. The interpretation of cardiac/cardiovascular disease is complex as these are late-onset, variable penetrance conditions. We have been collaborating with DECIPHER to implement features to enhance the interpretation of cardiac/cardiovascular disease, including the incorporation of additional data and providing a more intuitive experience for cardiac users, specifically when exploring the impact of a particular cardiac variant. In this blog post we describe how cardiac users can utilize the DECIPHER platform, with particular focus on these recent updates.
Flexible search bar
Within the cardiac community, different users (clinicians, diagnostic labs or researchers) may have their variant of interest formatted using a variety of positional identifiers. DECIPHER can now accept variants as search terms in a variety of formats.
Users can identify a gene by HGNC symbol, MANE select transcript or Ensembl identifier
Search bar can take the position within the gene as a coding DNA reference, protein reference, or using a “gnomad-style” search based on chromosome and genomic coordinates
Reference and alternate alleles can be identified using 1 or 3 letter amino acid codes
Additionally, the input bar allows for space or colon separators, and optional “p.” and “c.” prefix for cDNA and protein reference location
The result of incorporating these options allows for a wide range of search terms that a user can input to access data about a variant of interest. Most formatting used across the wider cardiac community is now accepted.
Many of these formats are highlighted under DECIPHER “Search examples” that appear when the search bar is accessed for entry.
Variant summary table
The landing page after entering a search term now leads with a summary that indicates whether the variant has been seen across multiple cohorts and datasets. Quick access to information about the presence of the variant in healthy or disease patients via DECIPHER patients, Deciphering Developmental Disorders (DDD) research variants, ClinVar, gnomAD, and other disease cohorts now including Cardiac VariantFX, can be informative as evidence towards a variant’s pathogenicity.
More information about the variant can be found by clicking on the summary link of a particular cohort.
Cardiac VariantFX Case/Control frequencies
Cardiac VariantFX has collected sequence and variant data from hypertrophic cardiomyopathy patients, dilated cardiomyopathy patients and healthy controls from a total of 7 centres*. Presence or absence of the variant in cardiac disease cohorts and/or confirmed healthy controls can be informative when assessing pathogenicity of a variant that is potentially associated with cardiac disease.
Only variants in genes known to be associated with hypertrophic or dilated cardiomyopathy at the time of processing are available on DECIPHER (ACTC1, BAG3, DES, DSP, FLNC, LMNA, MYBPC3, MYH7, MYL2, MYL3, PLN, RBM20, SCN5A, TNNC1, TNNI3, TNNT2, TPM1, TTN). Note that not all genes are sequenced in each cohort.
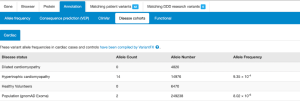
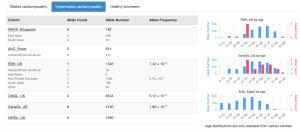
Cardiac VariantFX allele counts, allele numbers and calculated frequencies are accessible in DECIPHER’s “Annotation” tab under the “Disease cohorts” and “Cardiac” sub-tabs following a variant search. A tab with cardiac data is only shown when variant data is available for at least one of the cardiac cohorts. The upper panel of Disease Cohorts shows allele statistics as summed by disease status (HCM, DCM, healthy control, gnomAD exome and genome). The following variant counts are from the MYBPC variant ENST00000545968 p.Gln1233Ter.
The lower panel of Disease Cohorts breaks down each Cardiac VariantFX disease status by ethnicity (when available) and provides a histogram for the AC and AN by age for each cohort where age is available.
Gene-disease associations supported by cardiac cohort data
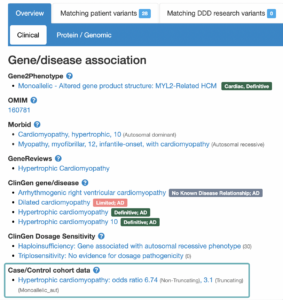
The landing page for each gene or variant search is a Gene-disease associations page (found below the variant summary table). For each gene, there are a number of sources (OMIM, PubMed Gene Reviews, Gene2Phenotype, ClinGen and GenCC) which have investigated conditions that are potentially associated. Associations listed on this page are not always definitive, and some investigations have concluded a lack of association.
In addition to the sources mentioned above, DECIPHER now also displays associations under a “Case/Control cohort data” heading, which now includes disease associations which have been supported by data included in the cardiac VariantFX cohort data.
Gene-disease association statistics
Clicking on the association supported by Case/Control cohort data opens a window that contains statistics that have been calculated based on cardiac cohorts** which support the association. Statistics are calculated individually for Truncating variants, Non-truncating variants, and All variants combined.
This window also contains a shortened text version of the mechanism narrative found in G2P. The full text version of the mechanism narrative can be found via the G2P VEP plugin.
Descriptions of each statistic can be found by clicking on the (“?”) found next to VariantFX gene-disease metrics. The current release contains statistics based on a subset of Cardiac VariantFX data which is also available at the Atlas of Cardiac Genetic Variation [https://www.cardiodb.org/acgv/]. Future releases will include updated statistics based on the full set of Cardiac VariantFX data, and also include an additional penetrance statistic.
Hot spot region (PM1) found in MYH7 visualised in the DECIPHER protein browser.
* Royal Brompton and Harefield Hospitals in London, UK, Aswan Heart Center in Egypt, National Heart Centre Singapore in Singapore, Laboratory for Molecular Medicine of Partners HealthCare Personalized Medicine in Boston, US, Oxford Regional Genetics Laboratory in the UK, GeneDx in Maryland, US, and Northern Ireland Regional Genetics Laboratory in Belfast, UK
** Excess in Disease, Aetiological Fraction, and Odds Ratio
Walsh , et al. Genet Med. 2017 Feb;19(2):192-203. doi: 10.1038/gim.2016.90. Epub 2016 Aug 17. PMID: 27532257; PMCID: PMC5116235.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, Ahmad M, Mazaika E, Allouba M, Zhang X, de Marvao A, Day SM, Ashley E, Colan SD, Michels M, Pereira AC, Jacoby D, Ho CY, Thomson KL, Watkins H, Barton PJR, Olivotto I, Cook SA, Ware JS. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019 Jan 29;11(1):5. doi: 10.1186/s13073-019-0616-z. PMID: 30696458; PMCID: PMC6350371.
Morales A, Kinnamon DD, Jordan E, Platt J, Vatta M, Dorschner MO, Starkey CA, Mead JO, Ai T, Burke W, Gastier-Foster J, Jarvik GP, Rehm HL, Nickerson DA, Hershberger RE; DCM Precision Medicine study of the DCM Consortium; Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med. 2020 Apr;13(2):e002480. doi: 10.1161/CIRCGEN.119.002480. Epub 2020 Mar 11. PMID: 32160020; PMCID: PMC8070981.
Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, Jongbloed JDH, Macaya D, Manrai A, Orland K, Richard G, Spoonamore K, Thomas M, Thomson K, Vincent LM, Walsh R, Watkins H, Whiffin N, Ingles J, van Tintelen JP, Semsarian C, Ware JS, Hershberger R, Funke B. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018 Mar;20(3):351-359. doi: 10.1038/gim.2017.218. Epub 2018 Jan 4. PMID: 29300372; PMCID: PMC5876064.
We are pleased to share our latest publication in Circulation on the genetic architecture of acute myocarditis and the overlap with inherited cardiomyopathy.
Myocarditis refers to inflammation of the heart muscle. It is traditionally considered to be a random event, which may be triggered by various infections, autoimmune conditions or toxic insults. Whilst most patients show spontaneous recovery, a small but important subset can suffer with long-term adverse cardiac events, including the need for heart transplantation or cardiac device implantation (1).
The purpose of this study was to answer a key question – is there an underlying genetic susceptibility that may explain the wide heterogeneity in clinical outcomes seen with acute myocarditis.
To address this, we compared 3 main cohorts:
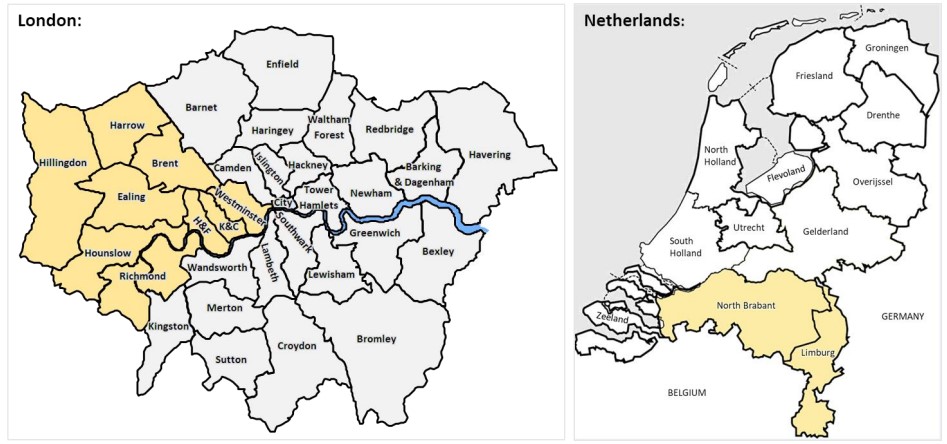
– Cohort 1: 230 patients recruited consecutively in London (UK) presenting with acute myocarditis confirmed on cardiovascular magnetic resonance (CMR) or myocardial biopsy (2).
– Cohort 2: 1053 community based healthy volunteers in London with no history of cardiovascular disease and a normal CMR scan.
– Cohort 3: 106 patients presenting with acute myocarditis confirmed on myocardial biopsy in Maastricht (Netherlands).
All participants underwent targeted DNA sequencing for well-characterized cardiomyopathy-associated genes with comparison to healthy controls sequenced on the same platform. The primary outcome was all-cause mortality.
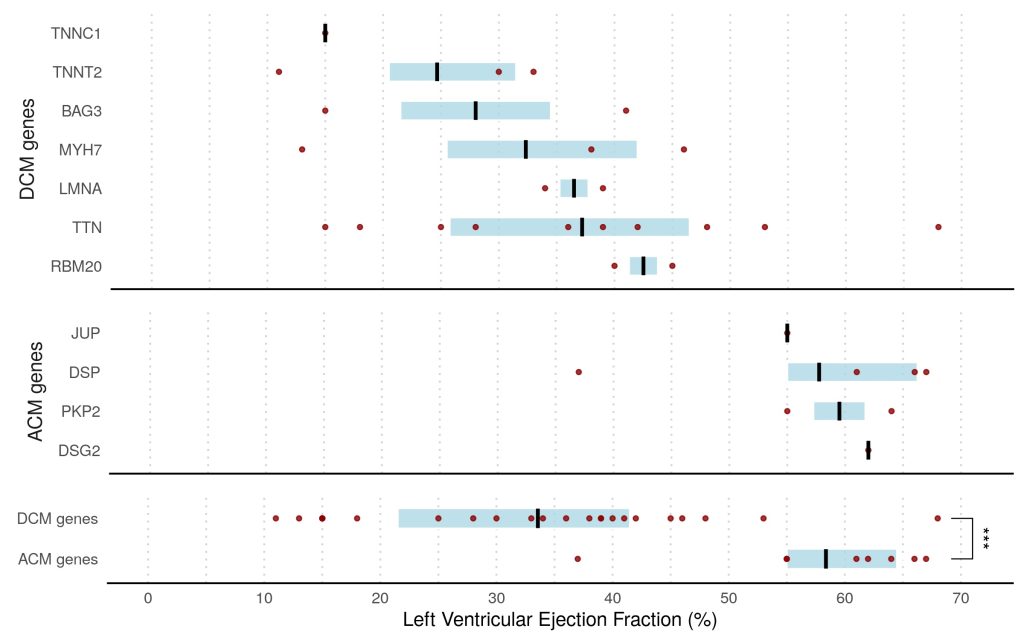
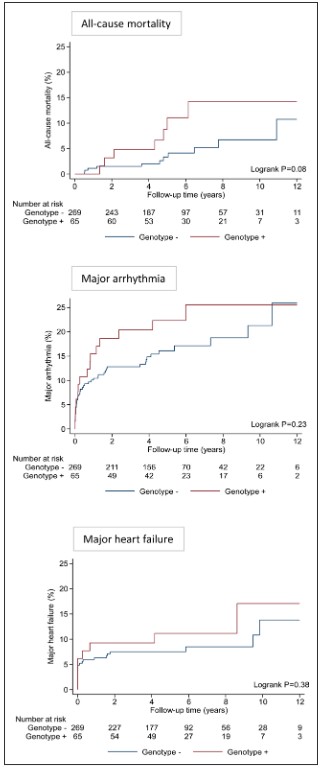
Overall, we found that 8% of patients presenting with acute myocarditis (27 out of 336 cases) carried likely pathogenic variants in ACM or DCM associated genes compared to <1% in healthy volunteers (p<0.0001).
This finding was dominated by truncating variants in Titin (TTN) in 7% of patients, all with left ventricular ejection fraction <50%, compared with 1% in controls (odds ratio, 3.6; P=0.0116). ACM-associated genes were found in 3% of cases versus 0.4% of controls (odds ratio, 8.2; P=0.001). This was driven predominantly by truncating variants in desmoplakin (DSP) in patients presenting with chest pain and preserved LV ejection fraction.
Over a median follow-up of 5.0 years (IQR, 3.9–7.8 years), there was a trend toward greater all-cause mortality in genotype-positive patients compared with genotype-negative patients (5-year mortality risk 11.1% vs 3.3%; P=0.08 after adjusting for age and sex).
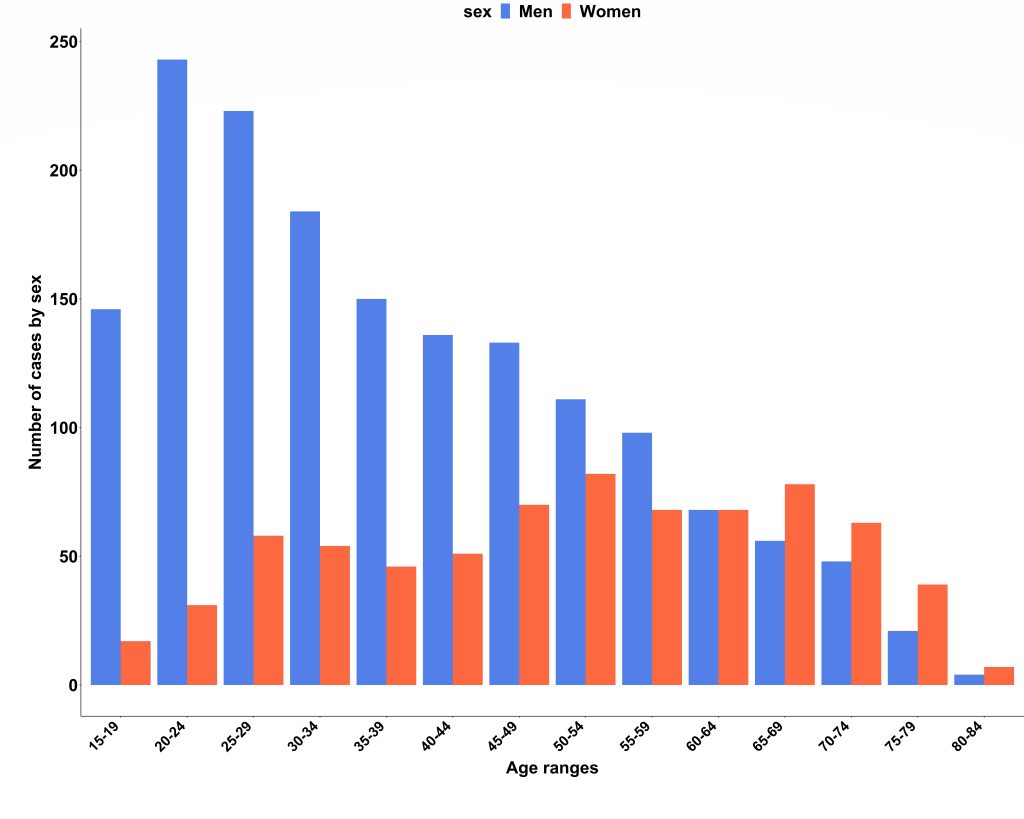
We obtained national hospital admission data from NHS Digital (3) and found there were 2353 admissions due to acute myocarditis over the 2-year study period (69% men; median age, 40 years; IQR, 27–55 years). Case ascertainment was calculated at 66%, and the London cohort was confirmed to be representative of national myocarditis admissions. Interestingly, we also found that men were significantly younger than women on admission to hospital (median age 35 years vs 52 years; P<0.001).
In conclusion, in this population-based study ~1 in 13 patients presenting with acute myocarditis were found to have an underlying variant in a gene robustly linked to DCM or ACM that would be reported as likely pathogenic in a patient with cardiomyopathy, compared with <1% in healthy controls. The presence of these variants affected clinical outcomes, particularly with DCM variants being associated with a trend towards greater all-cause mortality.
This study provides novel insights into why a small but important subset of patients with myocarditis experience major adverse events, whilst the majority usually recover spontaneously. It supports the concept that genotype-positive individuals may remain phenotypically silent until the occurrence of an environmental trigger.
This study suggests that genetic counselling and testing should be considered in patients with acute myocarditis, particularly in those with greater LV dysfunction, arrhythmia, or family history of cardiomyopathy. This may inform risk stratification and clinical management, including the need for ongoing surveillance and family screening, when cardiomyopathy-associated genetic variants are present.
A team of researchers with expertise across various disciplines was needed to harness the latest advances in precision phenotyping and genotyping whilst also leveraging national epidemiological datasets to deliver this study. Further work is underway to replicate these findings in a larger multi-centre setting and to provide greater mechanistic insights into genetic predisposition and disease progression in inflammatory cardiomyopathy using a systems biology approach.
The study was published under open access in Circulation on 26th Sept 2022:
Grun S, Schumm J, Greulich S, Wagner A, Schneider S, Bruder O, Kispert EM, Hill S, Ong P, Klingel K, et al. Long-term follow-up of biopsy-proven viral myocarditis: predictors of mortality and incomplete recovery. Journal of the American College of Cardiology. 2012;59:1604-1615
Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European heart journal. 2013;34:2636-2648, 2648a-2648d.